Clear Sky Science · en
Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome
When Breathing Fails Without Warning
Some children seem healthy at birth yet suddenly stop breathing during minor fevers, needing emergency ventilation. For their families, the episodes are terrifying and mysterious. This study investigates two such siblings from Japan and traces their life‑threatening spells of weakness and apnea (pauses in breathing) to tiny changes in a single gene that helps nerves talk to muscles. By piecing together clinical clues, gene sequencing, and computer‑based protein modeling, the researchers show how these mutations likely disrupt a key enzyme and give doctors a clearer target for diagnosis and treatment.
A Family Mystery of Sudden Weakness
The story centers on a brother and sister who both had slightly slow motor development in infancy. Around 18 months of age, each experienced episodes of apnea and loss of consciousness during fevers, serious enough to require a ventilator. As they grew, both continued to have bouts of drooping eyelids and generalized muscle weakness triggered by infections, fevers, or exertion. Brain scans were normal, and common antibody‑driven forms of myasthenia (an illness where communication between nerve and muscle is impaired) were ruled out. Yet a drug that boosts the chemical signal between nerves and muscles clearly improved their symptoms, pointing toward a rare inherited condition called congenital myasthenic syndrome.
Finding the Faulty Instructions
To look for an inherited cause, the team sequenced all protein‑coding genes in the siblings and their parents. They found that each child carried two different alterations in the same gene, CHAT, which encodes choline acetyltransferase—an enzyme that makes acetylcholine, the main chemical messenger used by nerves to activate muscles. One alteration changed a single building block of the enzyme (a missense mutation known as G411R). The other sat at a critical boundary where the cell normally cuts and joins gene segments while making RNA (a splice‑site mutation labeled c.752+2T>C). Each parent carried only one of these changes and was healthy; only the children who inherited both showed disease, suggesting that the pair of mutations together weakens enzyme function.
Probing How a Hidden Cut Alters the Enzyme
Because the researchers could not get enough natural CHAT RNA from blood cells, they used a "minigene" experiment. They cloned the relevant stretch of the gene into a DNA vector, introduced either the normal or mutated version into cultured cells, and then examined how the RNA was processed. In the normal construct, the RNA contained all expected segments. In the mutant version, an entire segment known as exon 5 was skipped, yet the overall reading frame of the gene remained intact. This meant that the enzyme would be made but missing a short internal stretch of amino acids. Evolutionary comparisons showed that this missing region is highly conserved across species, hinting that it plays an important structural role.
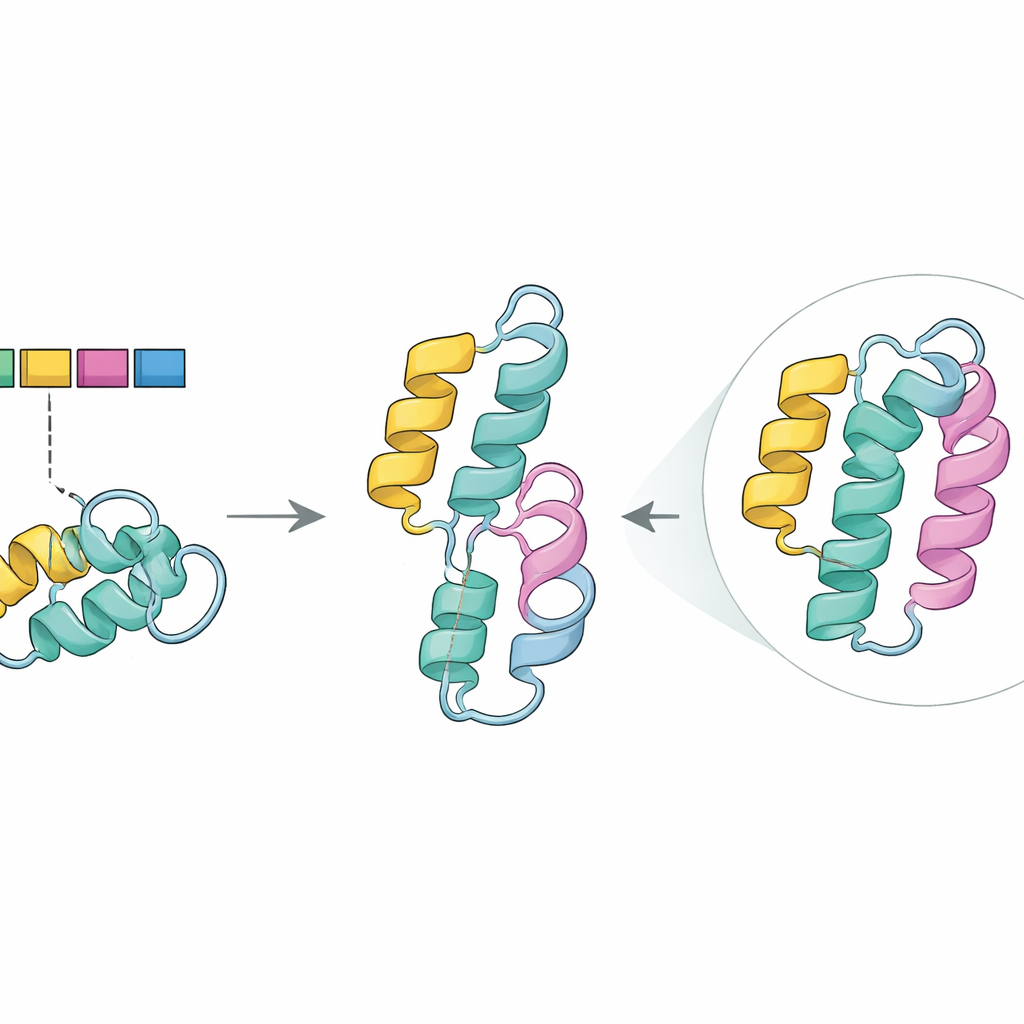
Seeing Structural Damage in Silico
To explore that role, the team turned to AlphaFold2, an advanced program that predicts three‑dimensional protein shapes from their sequences. In the normal enzyme, the portion encoded by exon 5 forms one of the tightly packed spiral segments (an alpha helix) that help stabilize the core of the protein. In the predicted mutant structure, this helix vanished, leaving a gap in a region known from earlier work to be crucial for maintaining stability and supporting efficient chemistry. Together with computer tools that flag damaging mutations, these results support the idea that skipping exon 5, especially when paired with the G411R change on the other copy of the gene, undermines the enzyme’s performance without completely eliminating it—consistent with the siblings’ moderate but serious symptoms.
What This Means for Patients and Families
The study concludes that the combination of the G411R missense mutation and the newly identified splice‑site mutation in CHAT is very likely responsible for the siblings’ congenital myasthenic syndrome. By demonstrating, through the minigene assay and structural modeling, how the splice‑site change removes a stabilizing helix from the enzyme, the authors provide a mechanistic explanation that clinicians and researchers can build on. For affected families, such work offers more than a name: it supports tailored treatment with drugs that boost neuromuscular signaling, guides genetic counseling for future pregnancies, and adds an important new example to the catalog of how subtle changes in our genetic code can profoundly influence muscle strength and the basic act of breathing.
Citation: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Keywords: congenital myasthenic syndrome, CHAT gene, choline acetyltransferase, splice-site mutation, neuromuscular junction