Clear Sky Science · en
Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction
Why this matters for future medicines
Designing a new medicine is a bit like designing an aircraft: you want to know how it will behave long before you build it for real. For drugs, that behavior includes how easily they evaporate, how they mix with water or fats, and how they move through the body. This article shows how carefully crafted mathematics can predict many of these physical and chemical traits from a drug’s structure alone, potentially saving time, cost, and trial‑and‑error in drug discovery. 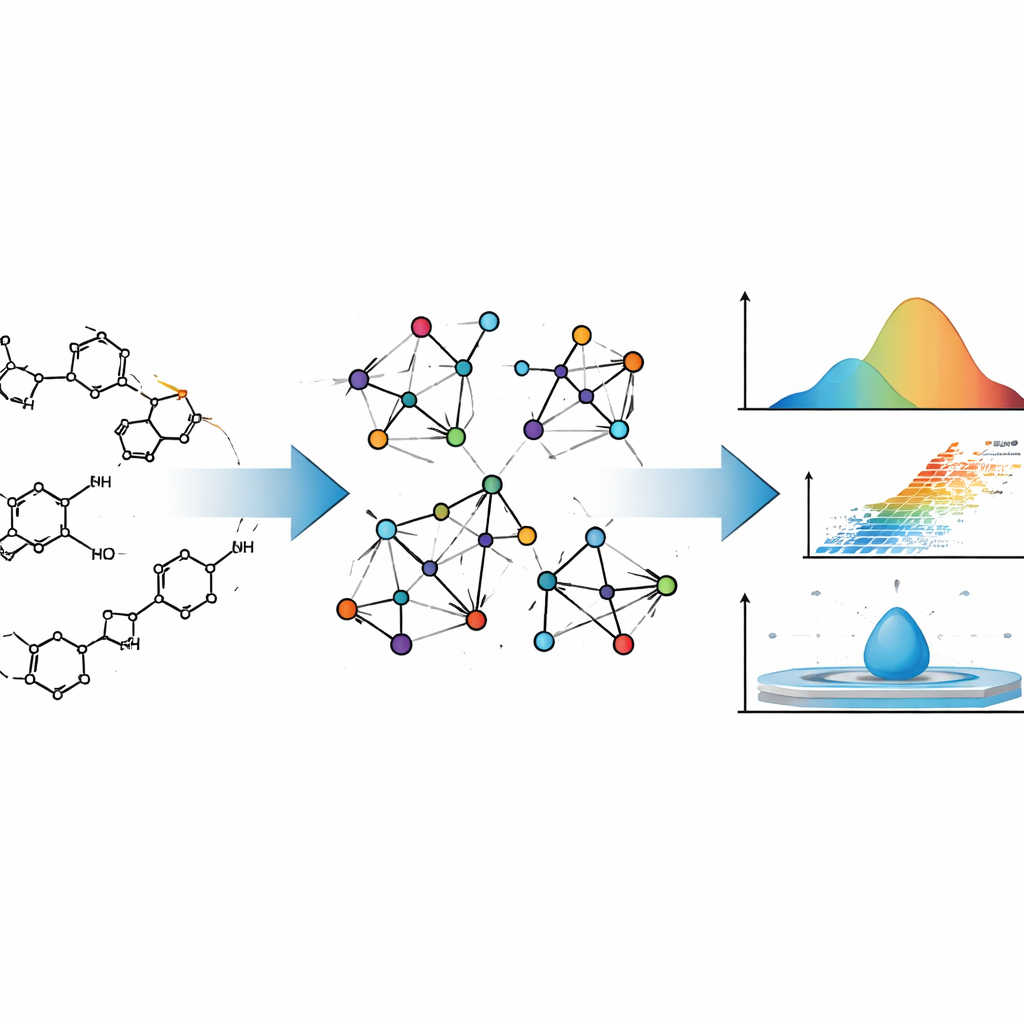
From molecules to networks
The authors treat drug molecules not just as collections of atoms, but as networks. In this picture, each atom is a point and each chemical bond is a line connecting two points. This kind of description comes from graph theory, a branch of mathematics that studies networks of all kinds, from social media links to power grids. Chemists have used such “molecular graphs” for decades, because certain numerical summaries of these graphs—called topological indices—often track how molecules behave in the real world, such as how easily they boil or how dense they are.
Adding neighborhood detail to the picture
Traditional indices usually pay attention only to how many bonds touch each atom. The team behind this study goes a step further. They use so‑called neighborhood M‑polynomial (NM‑polynomial) indices, which do not just count an atom’s own connections but also summarize how connected its neighbors are. This richer description captures subtleties such as how branched a molecule is, how its rings are fused together, and where oxygen or nitrogen atoms sit within the framework. Those features in turn influence how strongly molecules stick to each other, how rigid they are, and how their electrons respond to electric fields—all ingredients of key physicochemical properties.
Testing the idea on real cancer drugs
To ground their mathematics in reality, the authors first work out NM‑polynomial indices for two well‑known anticancer agents, Mitoxantrone and Doxorubicin. Both are complex, multi‑ring molecules used widely in chemotherapy. By translating their detailed chemical drawings into molecular graphs and then into NM‑polynomial indices, the authors show how the method systematically tracks structural changes across different “sizes” of these molecules. They then automate this process with a Python program, which takes a molecule’s connectivity (in the form of an adjacency matrix) and instantly returns the full set of indices, minimizing human error and speeding up calculations that would be tedious by hand. 
Training machines to read molecular fingerprints
Next, the researchers move beyond these two drugs to a broader collection of 45 polycyclic medicines, including common names such as acetaminophen, ibuprofen, and several modern targeted therapies. For each drug they compile nine NM‑polynomial indices and nine experimentally measured properties: complexity, boiling point, enthalpy of vaporization, flash point, molar refractivity, polarizability, surface tension, molar volume, and index of refraction. They then train several machine‑learning style regression models—Linear, Ridge, Lasso, and Elastic Net—to learn how combinations of indices map onto each property. Careful statistical safeguards are used throughout: they remove redundant inputs, standardize variables, perform repeated cross‑validation on 80% of the data, and test the final models on an untouched 20%.
What the numbers reveal
The models show that NM‑polynomial indices are particularly powerful for properties tied to how molecules pack and interact. For boiling point, enthalpy of vaporization, flash point, molar refractivity, polarizability, and molar volume, the best models reach very high correlation scores, meaning the predicted values track the experimental ones closely. Regularized methods such as Ridge and Elastic Net generally perform best, suggesting that gently constraining the models helps them focus on the most informative aspects of the indices. A correlation heatmap confirms that several indices—especially those related to overall connectivity and “neighborhood richness”—are strongly and consistently aligned with these properties across the 45‑drug panel.
Limits and room for improvement
Not every property cooperates. The index of refraction, which captures how light bends when entering a material, proves stubborn: the models struggle to do better than simple averages, and the NM‑polynomial indices show only weak correlations with it. Surface tension is moderately well captured, but not as strongly as the other traits. These gaps hint that some behaviors depend on features beyond two‑dimensional connectivity, such as three‑dimensional shape or subtle electronic effects. The authors suggest that future work could blend NM‑polynomial indices with quantum‑chemical or 3D descriptors to bridge this divide.
What this means for drug design
In plain terms, the study shows that sophisticated but well‑structured math can turn a static sketch of a molecule into a surprisingly accurate predictor of how that molecule behaves in the lab. For many important properties—how hard it is to boil, how bulky it is, or how easily its electrons shift—the NM‑polynomial approach, combined with modern regression techniques, matches or outperforms earlier methods that used simpler indices or smaller datasets. While it is not yet a complete replacement for experiments, it offers drug designers a faster screening tool: by computing these graph‑based fingerprints, they can estimate key physicochemical properties early on, focus laboratory work on the most promising candidates, and explore chemical space more efficiently.
Citation: Tawhari, Q.M., Naeem, M., Koam, A.N.A. et al. Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction. Sci Rep 16, 8136 (2026). https://doi.org/10.1038/s41598-026-39562-9
Keywords: chemical graph theory, drug property prediction, molecular topology, machine learning in chemistry, physicochemical descriptors