Clear Sky Science · en
Structural modeling and docking analysis of canonical and novel resistance-associated missense mutations in Sudanese Escherichia coli
Why this matters for everyday health
Antibiotic-resistant infections are no longer rare medical curiosities; they increasingly threaten routine treatments for urinary tract infections, surgery, and intensive care. This study looks closely at Escherichia coli bacteria from Sudan and asks a very specific question: how do tiny genetic changes in bacterial proteins reshape the way common antibiotics work? By using computer-based structural modeling instead of expensive lab experiments, the authors uncover hidden resistance patterns that standard tests and global databases may miss—especially in low-resource settings where resistance is rising fastest. 
Looking inside the bacteria’s toolbox
The researchers focused on "missense" mutations—single-letter DNA changes that swap one building block of a protein for another. They analyzed whole-genome sequences from 55 E. coli isolates collected in Sudan and zoomed in on bacterial proteins that are direct targets of major antibiotic classes, including fluoroquinolones, macrolides, and rifampicin. These targets include DNA-twisting enzymes (gyrase and topoisomerase IV), the protein-making ribosome, and the enzyme RNA polymerase. Out of 71 mutations found in these proteins, 19 were flagged by multiple prediction tools as likely to damage protein function, and strikingly, most of these appeared to be novel variations not yet cataloged in global resistance databases.
New trouble spots in familiar targets
Some of the most important changes clustered in a ribosomal protein called L22, which helps form the tunnel through which newly made proteins exit the ribosome. This region also doubles as a docking site for macrolide antibiotics such as erythromycin. The study identified a dense set of previously unreported L22 mutations, many within a single strain, that sit right along this tunnel and at contact points with ribosomal RNA. Computational analyses suggested that several of these changes destabilize the local structure or make it more flexible, potentially reshaping the tunnel so that macrolide molecules fit less snugly. At the same time, more familiar "canonical" resistance mutations appeared in DNA-processing proteins ParC and ParE and in RNA polymerase, confirming that Sudanese strains share some global resistance hallmarks while also harboring their own local twists.
How shape changes weaken antibiotic grip
The team went beyond sequence lists and asked how these mutations might alter the three-dimensional fit between antibiotics and their targets. Using molecular docking simulations, they compared how different drugs bound to normal and mutated proteins. For the topoisomerase IV protein ParC, key mutations near the drug contact site substantially weakened the predicted binding of the fluoroquinolone trovafloxacin, reflecting a looser grip at the enzyme–DNA–drug junction. In the related ParE protein, mutations modestly reduced the binding of novobiocin. In contrast, a novel mutation in the gyrase protein GyrA appeared to destabilize the enzyme’s structure without noticeably changing how tightly the fluoroquinolone moxifloxacin could bind, hinting that resistance can sometimes emerge by subtly disrupting enzyme performance rather than by simply ejecting the drug. 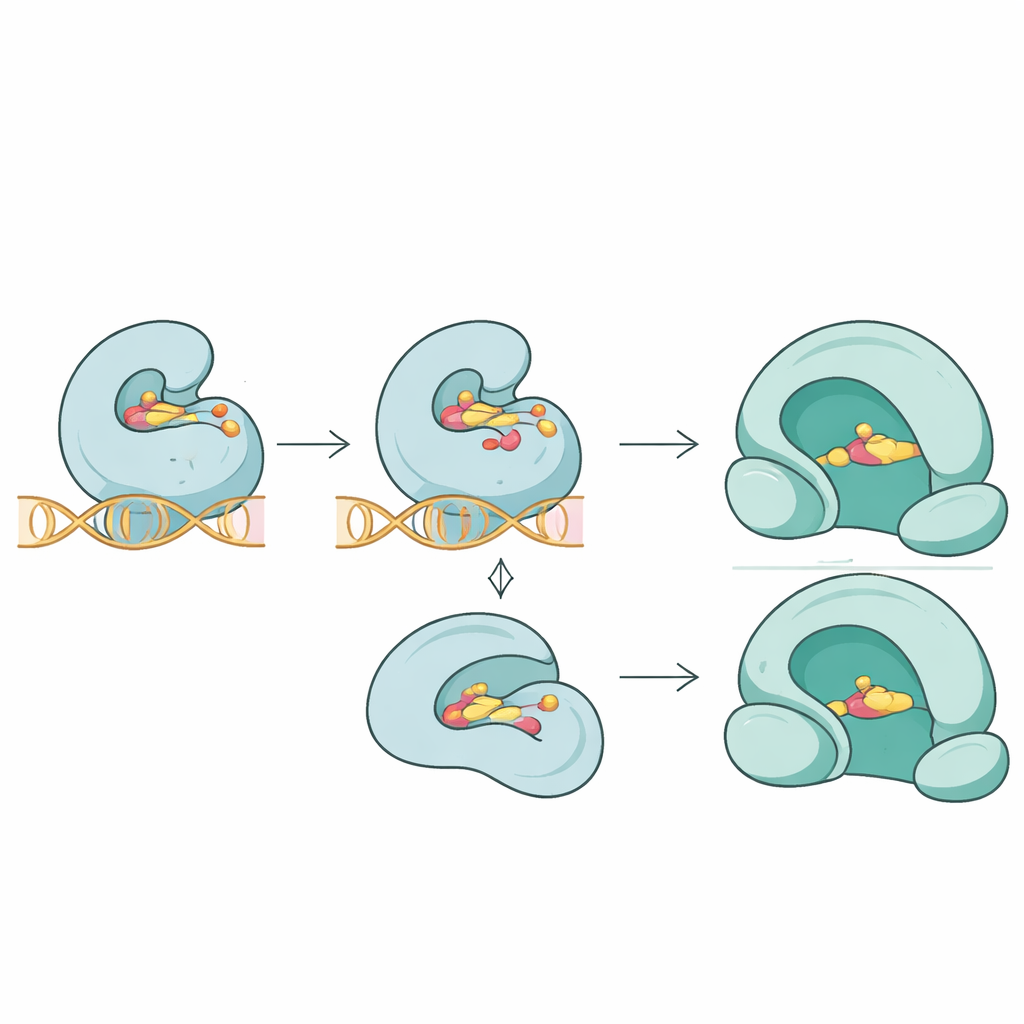
Mixed effects on different drugs
Not all mutations had the same impact. Classical rifampicin-resistance changes in the RNA polymerase protein RpoB did little to disturb the binding of a newer, structurally distinct inhibitor that targets a nearby site, suggesting that future drugs could be designed to bypass existing resistance patterns. For ribosomal protein L22, docking studies with erythromycin revealed a patchwork of outcomes: some mutations weakened binding, some had little effect, and one even slightly improved the predicted fit. These results emphasize that resistance is rarely all-or-nothing; instead, each mutation nudges protein stability, flexibility, and drug binding in different directions, and the overall effect on treatment depends on how these changes combine inside a living bacterial cell.
What this means for patients and surveillance
From a lay perspective, the key message is that bacteria in places like Sudan are evolving resistance through both well-known and lesser-known routes. The well-known routes involve classic mutations already tracked by international programs, but this study shows that many additional, locally enriched mutations may also weaken antibiotics in more subtle ways. By mapping these changes onto detailed protein structures, the authors provide a shortlist of mutations that should be tested in the lab and considered in regional diagnostic panels. In practical terms, their work argues that smart computer modeling can help countries with limited laboratory capacity keep better tabs on emerging resistance, ultimately supporting more reliable treatment choices and inspiring drug designs that stay one step ahead of bacterial evolution.
Citation: Sage, E.E., Ibrahim, S.A.E., Firdaus-Raih, M. et al. Structural modeling and docking analysis of canonical and novel resistance-associated missense mutations in Sudanese Escherichia coli. Sci Rep 16, 8995 (2026). https://doi.org/10.1038/s41598-026-39491-7
Keywords: antimicrobial resistance, Escherichia coli, missense mutations, structural bioinformatics, Sudan