Clear Sky Science · en
Integrating deep learning with physics based modeling enables high precision antibody antigen interface prediction
Why this matters for future medicines
Antibodies are the guided missiles of our immune system and of many modern drugs. To design better antibodies, scientists need to know exactly how an antibody grips its target molecule, or antigen. Measuring these structures experimentally is slow and expensive. This study shows how combining deep learning with classical physics-style modeling can sharply improve computer predictions of where an antibody and antigen touch, potentially speeding up antibody design and screening.
Finding the handshake zone
Antibodies recognize their targets using small flexible loops at their tips, called binding regions, which come together to form a contact patch. These loops can bend and twist, and the matching zone on the antigen is often spread out and shallow rather than forming a deep pocket. That flexibility and subtlety make the docking problem—figuring out how the two shapes fit together—extremely hard for computers. Traditional docking programs try out many relative positions of the two proteins and score them using physical rules such as electrostatic attraction and how water is displaced, but without any biological hints they often settle on incorrect matches.
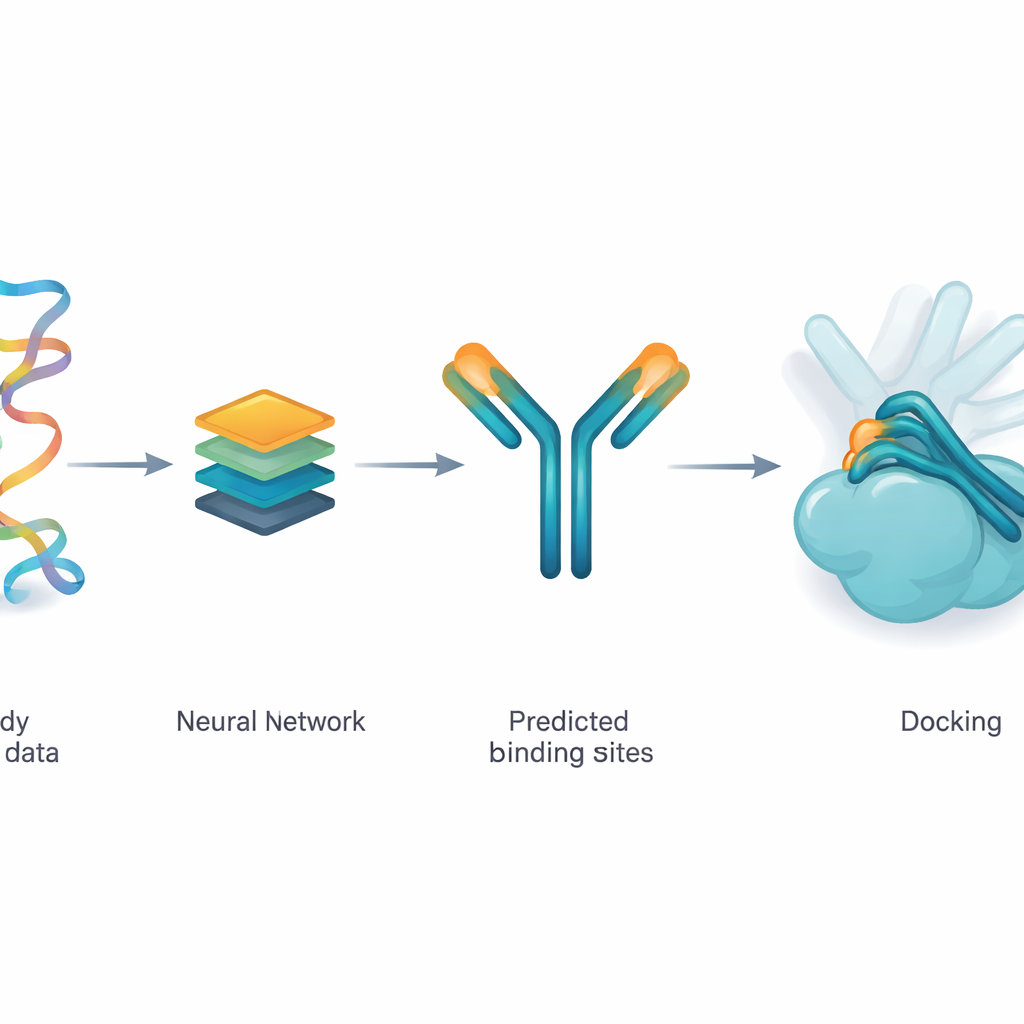
Teaching a network to suggest likely contact spots
The authors use a deep learning model called ParaDeep to guess which amino acids on an antibody are most likely to touch the antigen. ParaDeep does this using only the antibody’s sequence—the order of its building blocks—rather than needing a full 3D structure. It reads the heavy and light chain sequences together, encodes their chemical and positional features, and uses attention mechanisms to highlight residues that look like good candidates for binding. Each position receives a probability score; those above a threshold are treated as a predicted contact zone that can be mapped back onto the antibody structure.
Guiding a physics engine instead of replacing it
Rather than using deep learning to generate full antibody–antigen complexes from scratch, the team feeds ParaDeep’s predicted contact residues into an existing physics-based docking engine called PyDockWEB. This docking program samples thousands of possible ways the antibody and antigen could meet and scores them with an energy function. In the new framework, the predicted contact residues act as soft restraints: they bias the search so that many sampled orientations bring those residues near the antigen surface. Importantly, the underlying physical scoring and rigid-body treatment of the proteins are unchanged, making the process transparent and relatively lightweight to run.
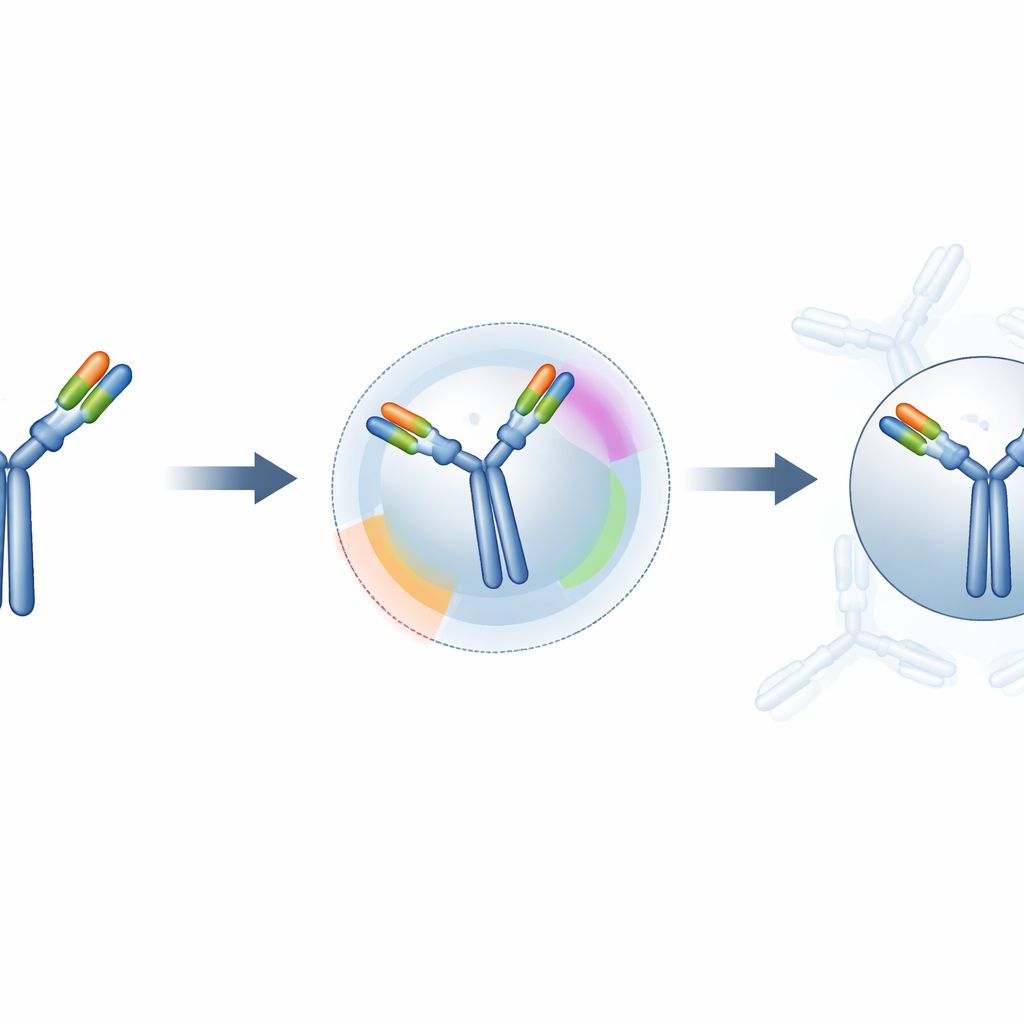
How much better do the predictions get?
The researchers tested their hybrid approach on 50 known antibody–antigen complexes from a curated database. For each case, they compared standard “blind” docking with docking guided by ParaDeep’s restraints. They measured local interface accuracy (how closely the predicted contact region matched reality), overall shape similarity, and a combined quality score that is widely used to rate docking models. Across this set, the guided method greatly reduced errors at the binding site, shifted overall structures closer to the true complexes, and moved many predictions from clearly wrong into medium or high quality categories. Nearly half of the guided models landed in the high-quality range, versus about a quarter for blind docking.
What makes some matches easier than others
The team also looked at why some complexes benefited more than others. They found that simply predicting more contact residues did not guarantee success; what mattered was placing restraints in the right area, not their number. Interfaces that were more water-loving and contained more flexible coil segments tended to dock better, likely because they worked well with PyDockWEB’s emphasis on electrostatics and were easier to align without major shape changes. When the researchers repeated some failed cases using “oracle” contact information extracted directly from experimental structures, most of those cases improved, confirming that accurate localization of the contact patch is a key ingredient—but rigid-body docking still has limits when large shape adjustments are needed.
What this means going forward
In everyday terms, this work shows that giving a physics-based docking program a smart hint about where an antibody is likely to grab its target can greatly improve its aim, without turning the process into an opaque black box. The combined ParaDeep–PyDockWEB pipeline does not replace more advanced flexible or generative methods, but offers a practical way to use sequence-level deep learning signals to guide familiar, interpretable docking tools. As antibody discovery and engineering efforts generate ever larger sequence libraries, such hybrid approaches could help researchers quickly filter candidates that are structurally consistent with a desired target, making the path from sequence to workable antibody faster and more informed.
Citation: Kodchakorn, K., Udomwong, P., Pamonsupornwichit, T. et al. Integrating deep learning with physics based modeling enables high precision antibody antigen interface prediction. Sci Rep 16, 8134 (2026). https://doi.org/10.1038/s41598-026-39466-8
Keywords: antibody docking, deep learning, paratope prediction, protein–protein interactions, antibody design