Clear Sky Science · en
FAM120A - a protein inserted in the ALS disease network
Why this matters for people and families
Amyotrophic lateral sclerosis (ALS) is a devastating disease that slowly paralyzes people by killing the nerve cells that control movement. Today, we still do not fully understand why these motor neurons die, and effective treatments remain scarce. This study shines a light on a little‑known protein, called FAM120A, and suggests it may help nerve cells cope with stress and prevent the buildup of harmful protein clumps—a hallmark of ALS. By uncovering how this protein behaves during disease, the work opens a fresh path toward understanding, and perhaps eventually treating, ALS.
Finding a hidden player in a crowded gene network
The researchers began not at the lab bench but at the computer. They used a “convergent analysis” approach to combine many existing datasets about ALS‑linked genes and their interactions. This network view allowed them to see clusters of proteins that work together in key cellular processes, especially those involving RNA handling and protein quality control—both known trouble spots in ALS. Within one such cluster, FAM120A emerged as a previously overlooked but highly connected protein that interacts with several established ALS‑related proteins. Its known roles in helping cells survive oxidative stress and in managing RNA made it a strong candidate for further study.
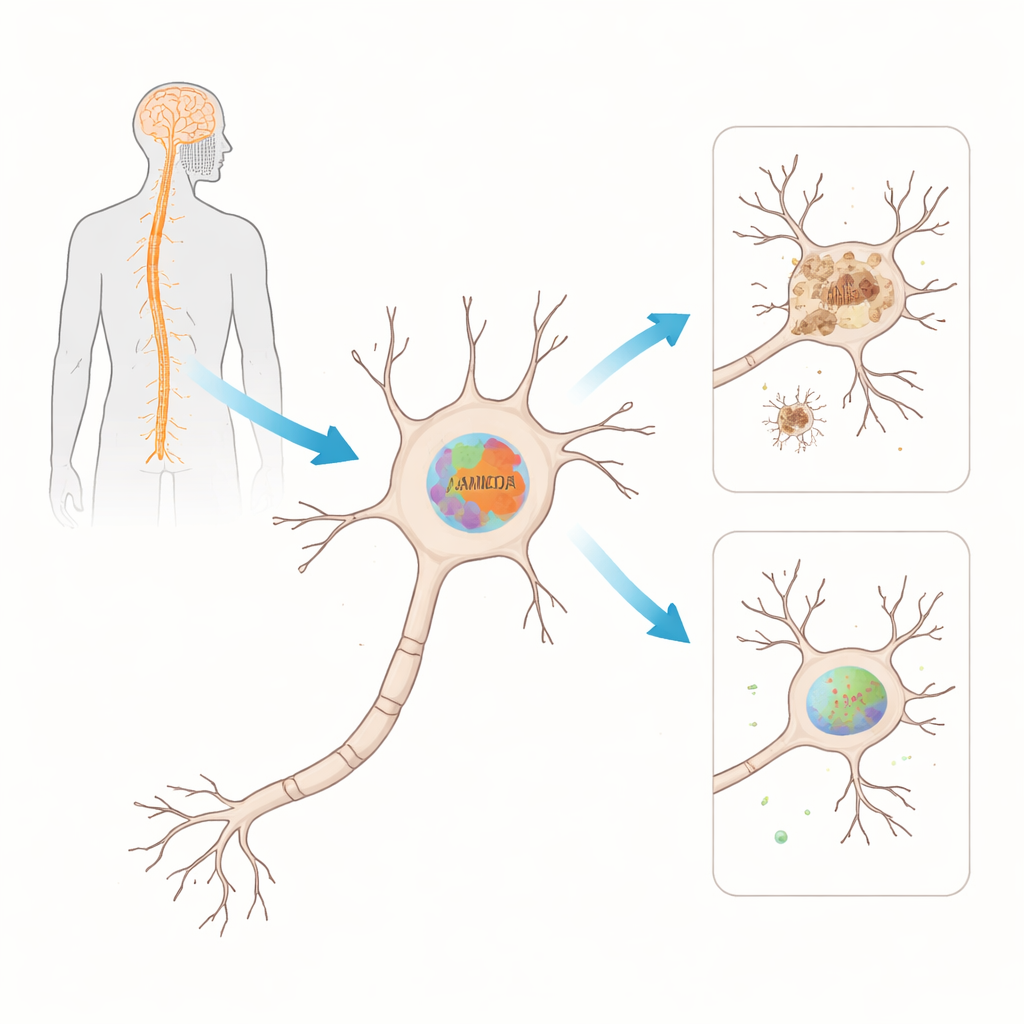
Tracking a vulnerable protein during disease progression
To test whether FAM120A really matters in ALS, the team turned to a widely used mouse model carrying a mutant version of the SOD1 gene, one of the first genetic causes identified in ALS. They measured both the RNA messages and the protein levels of the mouse version, Fam120A, in the spinal cord over time, from before symptoms appeared to late disease. Early on, the RNA levels of Fam120A dropped in the spinal cord before the animals showed clear signs of illness. Later, as paralysis developed, Fam120A protein itself was markedly reduced. This mismatch—RNA changing first, protein later—suggests that several layers of regulation break down as the disease advances.
Where in the spinal cord this protein lives
Next, the scientists asked where exactly Fam120A is found in the spinal cord. Using fluorescent microscopy on tissue slices, they observed that Fam120A is mainly present in neurons in the ventral horn—the region rich in motor neurons that degenerate in ALS. In late‑stage diseased animals, they saw some signal in support cells called astrocytes, but the dominant pattern remained neuronal. These observations link Fam120A directly to the very cells that fail in ALS and support the idea that its loss could weaken their ability to handle cellular stress, possibly contributing to motor function decline.
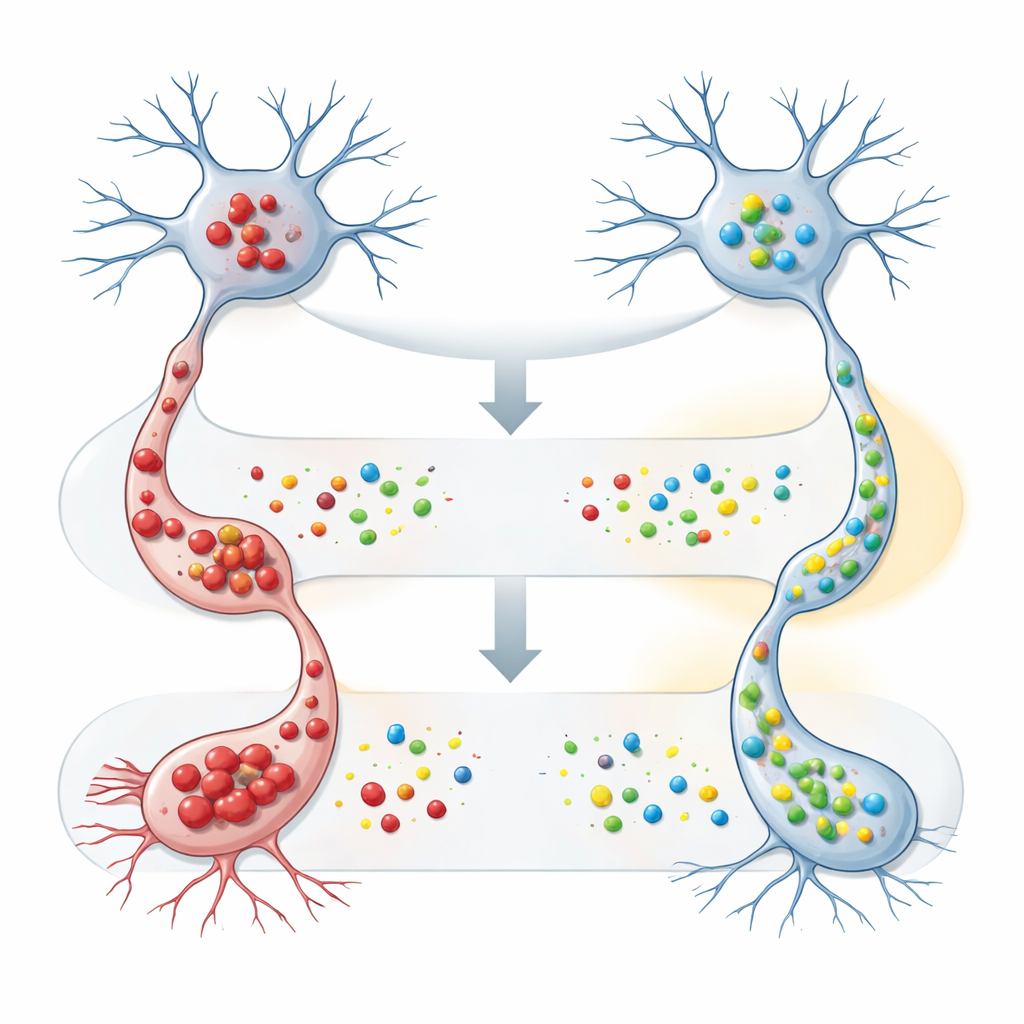
Putting extra FAM120A to work in nerve‑like cells
The team then moved to cultured nerve‑like cells to explore what FAM120A actually does. They engineered these cells to produce either normal or mutant SOD1, which tends to form toxic aggregates, and then forced the cells to make extra human FAM120A. When mutant SOD1 was present, boosting FAM120A significantly reduced both the amount of insoluble SOD1 detected by biochemical tests and the number of visible aggregates seen under the microscope. Importantly, FAM120A had little effect on the normal form of SOD1, hinting that it may specifically help cells manage misfolded or aggregation‑prone proteins, a central problem in ALS and other neurodegenerative diseases.
Building a broader map of molecular allies and enemies
Beyond these experiments, the researchers explored the wider interaction network of FAM120A. They confirmed that it physically associates with PURA, an RNA‑binding protein already linked to brain development and neurodegeneration, and found that PURA levels also fall in the ALS mouse spinal cord, though later in the disease. They highlight additional ties between FAM120A, its antisense partner gene FAM120Aos, and another RNA‑binding protein, ELAVL1, which regulates inflammatory and stress‑response genes in the brain. This growing web of connections places FAM120A at the crossroads of RNA regulation, stress responses, and protein quality control—precisely the systems that falter in ALS.
What this could mean for future ALS treatments
Taken together, the findings suggest that FAM120A is not just a bystander but a meaningful part of the ALS disease network. Its early decline in vulnerable motor neurons, its physical links to other RNA‑regulating proteins, and its ability to reduce toxic SOD1 clumps in cells all point toward a protective role in maintaining protein balance. While much work remains—especially to see whether similar changes occur in people with ALS and in other disease models—FAM120A now stands out as a promising target for future studies and, potentially, for therapies aimed at preserving the health of motor neurons.
Citation: Vicencio, E., Gomez, L., Beltran, S. et al. FAM120A - a protein inserted in the ALS disease network. Sci Rep 16, 8200 (2026). https://doi.org/10.1038/s41598-026-39329-2
Keywords: amyotrophic lateral sclerosis, motor neurons, protein aggregation, RNA-binding proteins, neurodegeneration