Clear Sky Science · en
Transcriptional activation of PPP1R14C by KLF7 unleashes CDK1 activity to promote lung squamous cell carcinoma
Why this lung cancer discovery matters
Lung cancer is still the leading cause of cancer deaths worldwide, and one major form—lung squamous cell carcinoma—has lagged behind in the era of targeted drugs. Unlike some other lung tumors that can be treated with medicines aimed at specific mutations, this subtype often forces doctors to rely on chemotherapy and immunotherapy, which do not work for everyone. This study uncovers a previously hidden control circuit inside lung squamous cancer cells that acts like cutting the brakes on cell division, and it points to a concrete weak spot that future drugs might exploit.
A missing link in a hard-to-treat lung cancer
The researchers began by searching large public cancer databases to see whether any genes stood out in lung squamous cell carcinoma. One, called PPP1R14C, consistently appeared at high levels in tumor samples compared with normal lung tissue. Its abundance rose as cancers advanced to later stages, and patients whose tumors made more of this molecule tended to live for a shorter time. These patterns held true both at the RNA level—the messages cells use to build proteins—and at the protein level itself, suggesting PPP1R14C was not just present but actively involved in driving the disease. 
How lung tumors release the brakes
To understand why PPP1R14C is so abundant in these tumors, the team turned to the gene’s on–off switch, its promoter. By combining several databases that track where different control proteins bind DNA, they zeroed in on a factor called KLF7 as a prime suspect. In lung squamous cancer cells grown in the lab, boosting KLF7 levels caused PPP1R14C to rise, while dialing KLF7 down sharply reduced it. Experiments that attached the PPP1R14C promoter to a light-emitting reporter confirmed that KLF7 could directly flip this switch; changing a short DNA sequence where KLF7 docks wiped out the effect. A technique that pulls down DNA bound to KLF7 from intact cells showed that this factor physically sits on the PPP1R14C promoter, sealing the case that KLF7 directly turns this gene on.
From genetic switch to aggressive behavior
Once they knew what pushes PPP1R14C up, the scientists asked what the molecule actually does. Using lung squamous cancer cell lines, they lowered PPP1R14C with genetic tools and watched the cells’ behavior. Cells lacking PPP1R14C grew more slowly, formed fewer colonies, invaded through a gel barrier less readily, and were more likely to undergo programmed cell death. The mirror image was also true: cells engineered to produce extra PPP1R14C divided faster, formed more colonies, and invaded more aggressively. When these altered cells were implanted into mice, tumors with reduced PPP1R14C grew smaller and weighed less. Together, these findings show that PPP1R14C is not a bystander but an active driver of cancerous traits.
A step-by-step look inside the cell-cycle engine
Digging deeper, the team examined which cellular programs depend on PPP1R14C. Broad surveys of gene activity revealed that removing PPP1R14C especially disrupted genes controlling the critical G2/M checkpoint—the point where a cell commits to splitting into two. At the heart of this checkpoint sits CDK1, a master switch for entry into mitosis. In cancer cells with high PPP1R14C, CDK1 carried an activating phosphate tag and its downstream targets lit up, signaling a green light for division. When PPP1R14C was reduced, this activation faded. Biochemical experiments showed why: PPP1R14C binds to a cellular “eraser” enzyme called PP1, which normally removes the activating tag from CDK1. By clamping onto PP1, PPP1R14C prevents it from reaching CDK1, so the activation signal persists and cells keep cycling. 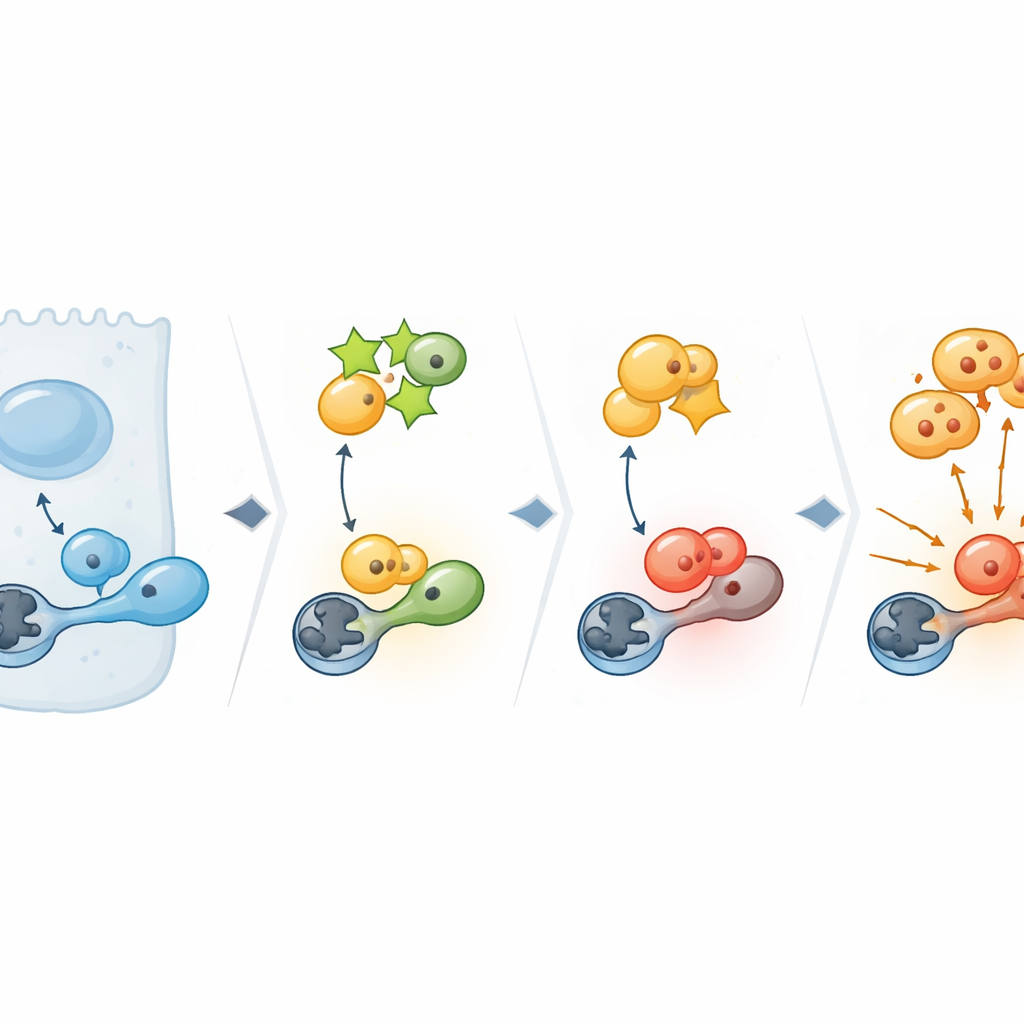
Turning a molecular insight into a treatment idea
The most encouraging part of the work came when the researchers tested a drug that directly blocks CDK1. In cells overloaded with PPP1R14C, this CDK1 inhibitor wiped out the growth advantage, reduced colony formation, and curtailed invasion, effectively reapplying the brakes that PPP1R14C had released. Putting the pieces together, the study outlines a clear chain of events: KLF7 switches on PPP1R14C; PPP1R14C neutralizes PP1; CDK1 remains hyperactive; and lung squamous cells divide unchecked. For non-specialists, this means scientists have identified both a warning flag—high PPP1R14C that marks more dangerous tumors—and a promising lever for therapy: drugs that shut down CDK1, particularly in patients whose tumors depend on this runaway circuit.
Citation: Xing, L., Yuan, C., Shen, X. et al. Transcriptional activation of PPP1R14C by KLF7 unleashes CDK1 activity to promote lung squamous cell carcinoma. Sci Rep 16, 9244 (2026). https://doi.org/10.1038/s41598-026-39174-3
Keywords: lung squamous cell carcinoma, cell cycle, CDK1, PPP1R14C, targeted therapy