Clear Sky Science · en
A2-pancortins interact with Bcl-xL and WAVE1 to promote mitochondria-ER contact sites (MERCs) and exacerbate mitochondrial calcium elevation to mediate cell death in stroke
Why tiny brain links matter in newborn stroke
Stroke is often thought of as an adult disease, but when it strikes newborns it can quietly damage the developing brain and cause lifelong problems. This study digs into what happens deep inside nerve cells during such a crisis. The authors focus on a little-known family of proteins called pancortins and show how two developmental versions act like dangerous “boosters,” tightening the connections between key cell structures and driving a flood of calcium that can push young neurons toward death.
Hidden culprits in the newborn brain
Pancortins are scaffold-like proteins that help shape the growing brain. Two forms, called A2-pancortins, are abundant during early development and then largely fade as the brain matures. Because newborn brains are especially vulnerable to oxygen loss, the researchers asked whether these early-life proteins might worsen damage when blood flow to the cortex is briefly cut off, as happens in neonatal stroke. In cultured mouse cortical neurons, they used genetic tools to lower all pancortin levels and then exposed the cells to an oxygen-and-glucose shortage that mimics stroke. Neurons with reduced pancortin were far more likely to survive, suggesting that these proteins, instead of protecting young cells, help drive them toward injury under stress.
From mouse stroke models to rescued brain tissue
To see whether this harmful role also appears in a living brain, the team engineered mice that specifically lack the developmental A2-pancortin forms. Young, two-week-old knockout mice and their normal littermates were then subjected to blockage of a major brain artery, a standard model of ischemic stroke. After one day, both groups showed injury in deep brain regions, but the cortex of mice lacking A2-pancortins had about half as much damage as that of normal animals. Strikingly, this protective effect vanished in five-week-old mice, when adult pancortin variants predominate. These age-dependent results point to A2-pancortins as key pro-death factors in the neonatal cortex, linking a developmental program to stroke vulnerability.
Dangerous contact zones inside neurons
Inside cells, the energy-producing mitochondria sit close to the endoplasmic reticulum (ER), a folded membrane network that stores calcium. Where the two membranes come into near contact, so-called mitochondria–ER contact sites act as microscopic tunnels through which calcium can pass. Moderate transfer supports energy production, but too much can overload mitochondria and trigger cell death. The researchers found that A2-pancortins, together with two partners—Bcl-xL and WAVE1—assemble into a three-part complex that sits at these contact sites. When they forced cells to make extra A2-pancortins plus these partners, mitochondria and ER touched more often and more tightly, as reported by a specialized split-fluorescent sensor. A tethering protein called GRP75 joined this complex, helping to stabilize the contact zones.
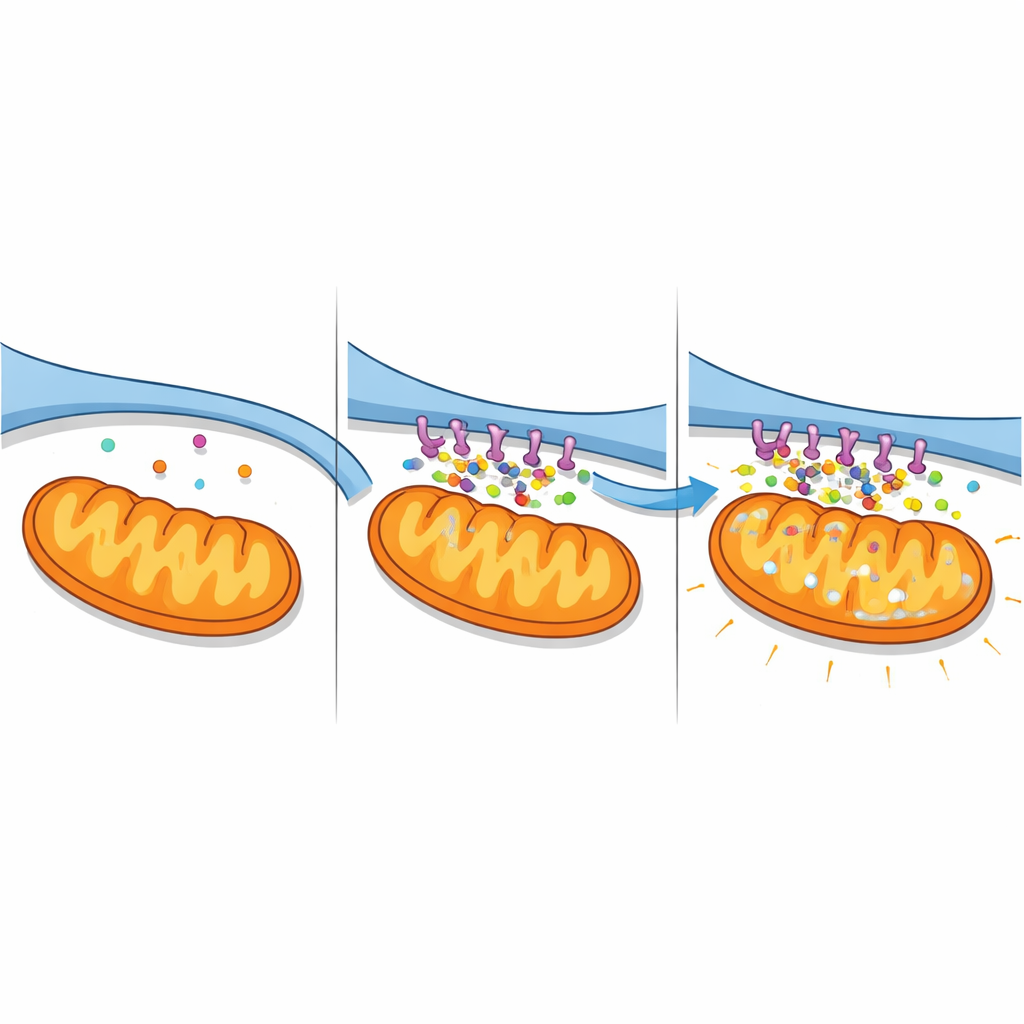
Calcium floods and failing power plants
The strengthened contacts had serious consequences for calcium balance. Using fluorescent indicators that separately report calcium in the cytosol, ER, and mitochondria, the authors watched levels change over time. Cells expressing the A2-pancortin complex showed a steady rise in calcium inside mitochondria and in the surrounding fluid, along with a drop in ER stores, a signature of massive transfer from ER into mitochondria. Blocking a key calcium-release channel on the ER surface (IP3R) largely prevented these changes, confirming that the complex amplifies a specific ER-to-mitochondria route. In nerve-like cells exposed to stroke-like oxygen and glucose deprivation, knocking down pancortins had the opposite effect: calcium overload was blunted and ER stores were better preserved. Together, these findings reveal A2-pancortins as organizers of a calcium “super-highway” that becomes deadly under ischemic stress.
What this means for protecting newborn brains
For non-specialists, the take-home message is that this work identifies a new molecular switch that helps decide whether young neurons live or die after stroke. By tightening the microscopic junctions between calcium-storing membranes and cellular power plants, A2-pancortins cause mitochondria to choke on excess calcium and fail. Removing these proteins in young mice softens the impact of experimental stroke, hinting that drugs or gene-based therapies aimed at disrupting A2-pancortin complexes—or loosening the contact sites they reinforce—could one day reduce brain injury in affected newborns. While such treatments remain future prospects, the study maps a clear and testable pathway from developmental proteins to calcium overload and neuronal loss in neonatal stroke.
Citation: Yang, Q., Wang, CC., Matsuyama, T. et al. A2-pancortins interact with Bcl-xL and WAVE1 to promote mitochondria-ER contact sites (MERCs) and exacerbate mitochondrial calcium elevation to mediate cell death in stroke. Sci Rep 16, 8467 (2026). https://doi.org/10.1038/s41598-026-38928-3
Keywords: neonatal stroke, mitochondria-ER contact sites, calcium overload, pancortin proteins, neuronal cell death