Clear Sky Science · en
Conformational dynamics and binding free energy analyses unveil a stable flavonoid inhibitor of dengue virus NS5 polymerase
Why a mosquito-borne virus needs plant-based help
Dengue fever has exploded worldwide in recent years, sickening millions and killing thousands. Yet we still lack a widely effective, affordable antiviral pill to treat people once they are infected. This study asks a deceptively simple question with big implications: can naturally occurring plant chemicals be turned into precise molecular "wrenches" that jam one of dengue virus’s most important machines?
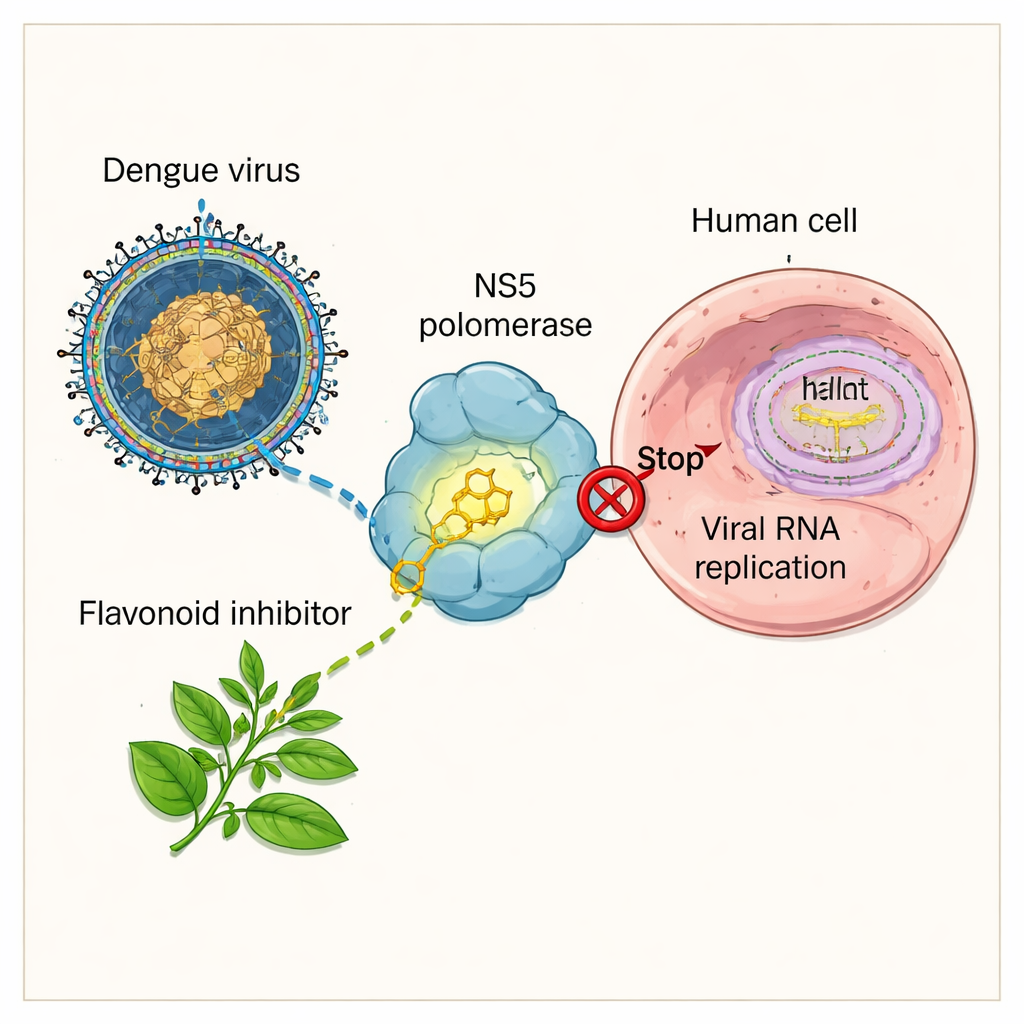
The virus engine scientists aim to shut down
Dengue virus survives by copying its genetic material inside our cells. To do this, it relies on a key protein called NS5, which acts like a tiny copying engine for the virus’s RNA. If NS5 stops working, the virus cannot make new genomes and the infection stalls. Drug developers therefore see NS5—especially its RNA-copying region, known as an RNA-dependent RNA polymerase—as a prime target for new medicines. Several synthetic chemicals and plant extracts have already shown that NS5 can be blocked, but many of these early candidates either bind weakly, are unstable, or raise concerns about safety and how well they move through the body.
Searching plants for promising chemical needles in a haystack
The researchers focused on flavonoids, a large family of plant compounds found in foods like berries, tea, and herbs, long known for antiviral and anti-inflammatory effects. From a curated database of plant secondary compounds, they pulled out 326 flavonoids and prepared three-dimensional models of both the virus’s NS5 enzyme and each candidate molecule. Using stepwise computer docking, they asked: which shapes fit best into a specific “backdoor” region on NS5 called the N-pocket—an allosteric site that can switch the enzyme off when occupied, and that tends to mutate less than the main active site?
At each stage of this virtual screening, compounds that fit poorly or looked unlikely to behave like real drugs in the body were discarded. The remaining molecules were then scored not just on geometric fit, but also on estimated binding strength using a method that approximates the energies involved when a compound sticks to NS5 in watery, cell-like conditions. This process narrowed the field to a handful of top flavonoids, with one candidate, labeled PSCdb01560, standing out for its particularly strong predicted binding energy.
Watching molecules move in a virtual microscope
Fitting well in a static snapshot is not enough; a useful drug must also stay put when everything jiggles in real life. To test this, the team ran long, detailed molecular dynamics simulations—computer movies lasting the equivalent of half a microsecond—for NS5 paired with the best flavonoids and a known reference inhibitor. They tracked how much the protein and each compound wobbled, how tightly packed the complex remained, how exposed it was to surrounding water, and how often key chemical contacts formed and broke. PSCdb01560 showed remarkably steady behavior: it stayed deeply seated in the N-pocket, moved very little compared with rival molecules, and seemed to stabilize the overall shape of NS5 rather than disturb it.
By contrast, two other flavonoids that initially looked promising began to drift within the pocket or became more exposed to the solvent as time went on—signs of weaker, less reliable binding. When the researchers mapped the “free energy landscape” of each complex—a way of visualizing which shapes are thermodynamically favored—they found that PSCdb01560 occupied a deep, well-defined energy valley, while the less stable compounds hopped among several shallower basins. When they compared the lowest-energy snapshots back to the original docked poses, PSCdb01560’s position hardly changed, underscoring its conformational loyalty.
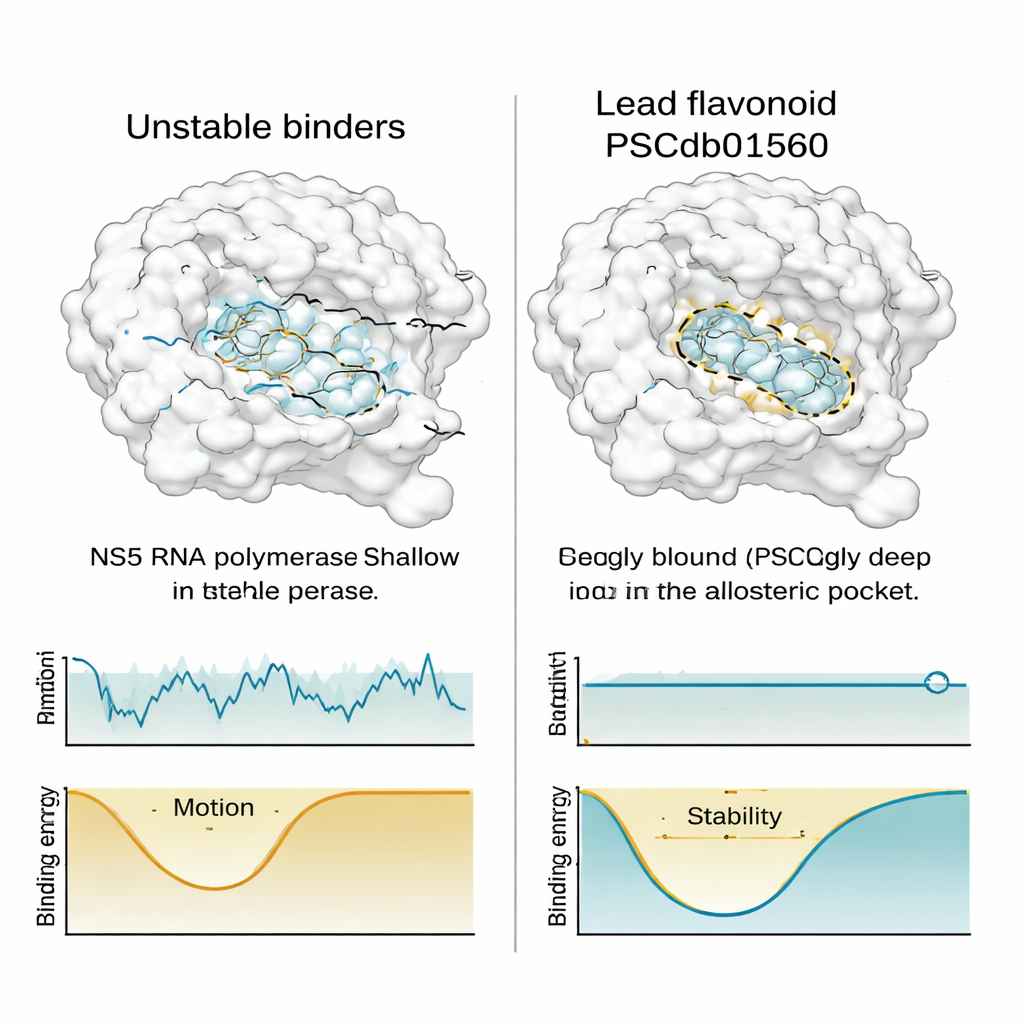
Numbers that hint at drug-level strength
Finally, the team used an energy calculation framework to estimate how strongly each flavonoid would bind after all the simulated motion was taken into account. PSCdb01560 achieved a binding free energy more favorable than the established reference compound, driven by a combination of snug shape complementarity, attractive electrical interactions, and oil-like contacts inside the pocket. This pattern—strong computed affinity, stable positioning over time, limited internal flexing, and engagement of crucial NS5 residues shared across dengue strains—marks PSCdb01560 as a particularly compelling starting point for drug design.
What this could mean for future dengue treatments
These results do not yet deliver a pill for dengue, but they significantly narrow the search. The work identifies one plant-derived flavonoid scaffold that, on the computer, outperforms several competitors and even a known benchmark inhibitor in both stability and predicted binding strength. The next steps are experimental: testing whether PSCdb01560 really blocks NS5 in test tubes, stops dengue from replicating in infected cells, and proves safe in animal models. If those hurdles are cleared, chemists could fine-tune this flavonoid into a clinically useful antiviral. For now, the study offers a hopeful message: nature’s own chemical library still holds promising tools for disarming one of the world’s fastest-growing mosquito-borne threats.
Citation: Alsaady, I.M., Gattan, H.S., Aljahdali, S.M. et al. Conformational dynamics and binding free energy analyses unveil a stable flavonoid inhibitor of dengue virus NS5 polymerase. Sci Rep 16, 7761 (2026). https://doi.org/10.1038/s41598-026-38864-2
Keywords: dengue virus, NS5 polymerase, flavonoid inhibitor, antiviral drug discovery, molecular docking