Clear Sky Science · en
Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis
Why making plant proteins behave matters
Many of the proteins that control how plants grow are large, fragile molecules that refuse to dissolve when scientists try to study them in the lab. One such protein, called DEK1, helps shape plant bodies from the level of single cells upward. But because a crucial part of DEK1 clumps together when produced in bacteria, its 3D structure has remained unknown, slowing efforts to understand and harness it. This study shows how computer modelling and smart, data-driven design can redesign that troublesome region to be more soluble, without breaking how it is built—offering a general recipe for taming difficult proteins.
Targeting the trouble spot in a key plant protein
DEK1 is an unusually large protein embedded in cell membranes and capped by a cutting enzyme region known as a calpain domain. Genetic work has shown that this domain is essential for normal development in plants such as mosses and crops, yet its structure has never been solved experimentally. When researchers try to make this calpain core (called CysPc) in the common host bacterium Escherichia coli, it tends to become insoluble and forms dense inclusion bodies. That makes it nearly impossible to purify in the amounts and quality needed for detailed structural and functional studies. The authors therefore set out to redesign the CysPc domain so that it would dissolve more easily while preserving its overall shape.
Building a trustworthy 3D model from scratch
Because no experimental structure exists for this plant calpain, the team first had to predict its 3D form. They combined several state-of-the-art structure prediction tools, including AlphaFold2, SWISS-MODEL and I-TASSER, and anchored these predictions to known structures of related mammalian calpains. Using a consensus approach, they refined and checked the resulting models with multiple quality tests that assess backbone geometry, packing, and agreement with known structural patterns. These independent checks showed that the integrated model of the CysPc domain was more reliable than any single prediction alone, providing a solid starting point for exploring how small changes to the amino-acid sequence might improve solubility.
Testing virtual mutations in a simulated solvent
With the 3D model in hand, the authors ran extensive molecular dynamics simulations, in which the protein and surrounding water molecules are followed over time on the computer. They focused on residues on the protein surface that were flexible, hydrophobic, or predicted to promote aggregation. Candidate positions were mutated individually to more water-friendly amino acids and then simulated for 200 nanoseconds each. For every variant they measured features related to solubility, such as how much surface area contacts water, how compact the protein remains, and how strongly atoms fluctuate. Many single mutations modestly increased solvent exposure or internal hydrogen bonding while leaving the overall fold unchanged, suggesting that the basic scaffold of CysPc could tolerate carefully chosen substitutions.
Letting algorithms search the mutation space
Changing just one residue rarely produces dramatic gains in solubility, so the researchers next explored combinations of two and three mutations. They generated a library of double and triple variants built from the best single mutations and again simulated each one. To score and rank these designs fairly, they defined a weighted index that combines multiple simulation features known to correlate with solubility, rewarding increased hydration and internal bonding while penalizing excessive flexibility. They then used a reinforcement learning algorithm (Proximal Policy Optimization) to navigate the huge space of possible triple mutants and propose the most promising combinations. This data-driven search converged on a particular triple mutant, named MUT347, as the top candidate.
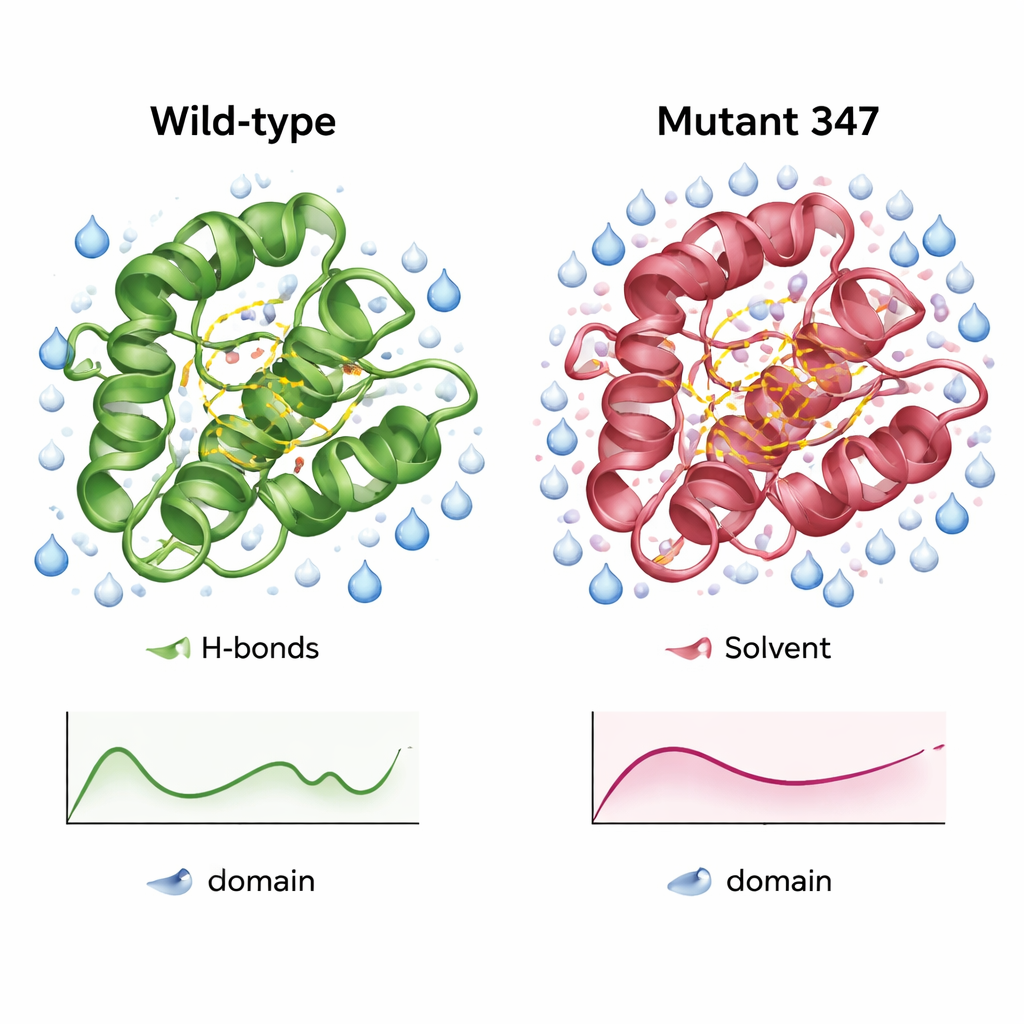
A more compact, better-hydrated version of the enzyme
Detailed simulations of the wild-type CysPc domain and MUT347 revealed how the engineered variant differed. MUT347 equilibrated more quickly and showed smaller overall deviations from its starting shape, indicating greater structural stability in solution. Its loops and chain ends were slightly less floppy, while the core catalytic region retained its original flexibility, suggesting that functionally important motions were preserved. The triple mutant had more internal hydrogen bonds and a larger water-accessible surface in key regions, signs of a better organized and more hydrated surface. Under varying salt concentrations and pH levels, MUT347 consistently maintained lower fluctuations than the original protein, behavior associated with reduced tendency to clump.
What this means for studying and reusing proteins
For non-specialists, the takeaway is that the authors have built a largely computer-based recipe to turn an awkward, clumping piece of a vital plant protein into a more soluble, well-behaved version, without having to know its structure beforehand from experiments. By combining modern structure prediction, long-timescale simulations, and learning algorithms that can juggle many design choices at once, they identified a triple mutation that is predicted to stabilize the fold and expose it more favorably to water. While experimental work is still needed to confirm the gains in real test tubes, this framework could be broadly useful for rescuing other eukaryotic proteins that are hard to produce, ultimately helping scientists unlock structures and functions that are currently out of reach.
Citation: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Keywords: protein solubility, computational mutagenesis, molecular dynamics, plant calpain DEK1, protein engineering