Clear Sky Science · en
LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling
Why heart scarring matters
Heart failure affects tens of millions of people worldwide and often creeps up silently over years. A major driver of this decline is myocardial fibrosis—slow, progressive scarring of the heart muscle that makes it stiffer and less able to pump blood. This study digs into the molecular "wiring" that tells heart cells to lay down too much scar tissue, and identifies a new chain of molecules that could be targeted to slow or even reverse this harmful process.
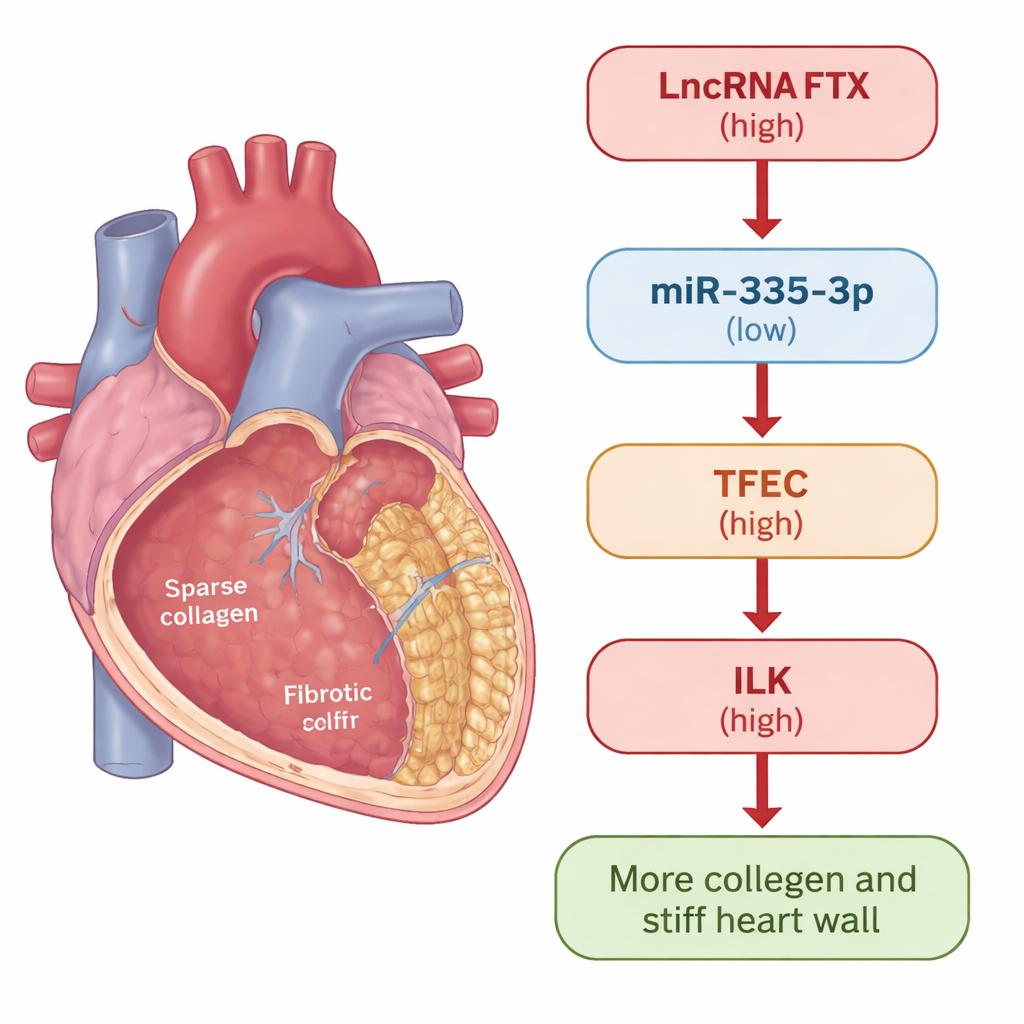
A closer look at heart scarring
When the heart is injured or stressed, support cells called cardiac fibroblasts spring into action. In healthy repair, they help patch damage. But in chronic disease they can switch into an overactive state, churning out excess collagen and other components of the extracellular matrix, ultimately stiffening the heart wall. The researchers used two models to study this process: mice treated with the drug isoproterenol, which reliably induces heart fibrosis, and human cardiac fibroblasts exposed to the signaling molecule TGF-β1, a well-known trigger of scarring. In both settings, they measured how specific genes and proteins changed as fibrosis developed.
A harmful chain reaction inside cells
The team focused on a transcription factor called TFEC, a protein that sits in the cell nucleus and turns other genes on. They found that TFEC, along with another protein called integrin-linked kinase (ILK), was consistently increased when fibroblasts were pushed toward a fibrotic, scar-forming state. Silencing either TFEC or ILK sharply reduced classic fibrosis markers such as α-smooth muscle actin and collagens I and III, and also dialed down a growth-control pathway (Akt/GSK3β and Hippo signaling) known to promote tissue scarring. Experiments mapping DNA binding showed that TFEC directly attaches to the promoter of the ILK gene and boosts its activity, placing TFEC clearly upstream of ILK in a pro-fibrosis signaling cascade.
RNA switches that control the master regulator
To understand what controls TFEC itself, the researchers turned to noncoding RNAs—RNA molecules that do not make proteins but act as fine-tuners of gene activity. They identified a small RNA, miR‑335‑3p, that was reduced in fibrotic hearts and cells. Raising miR‑335‑3p levels lowered TFEC, while blocking it increased TFEC, and reporter tests confirmed miR‑335‑3p binds directly to TFEC messages to keep them in check. They then found a long noncoding RNA, called FTX, that was elevated in fibrosis and physically interacted with miR‑335‑3p. FTX acted like a molecular sponge: it soaked up miR‑335‑3p, preventing this small RNA from restraining TFEC. As a result, TFEC and ILK rose, and fibroblasts produced more scar-forming collagen.
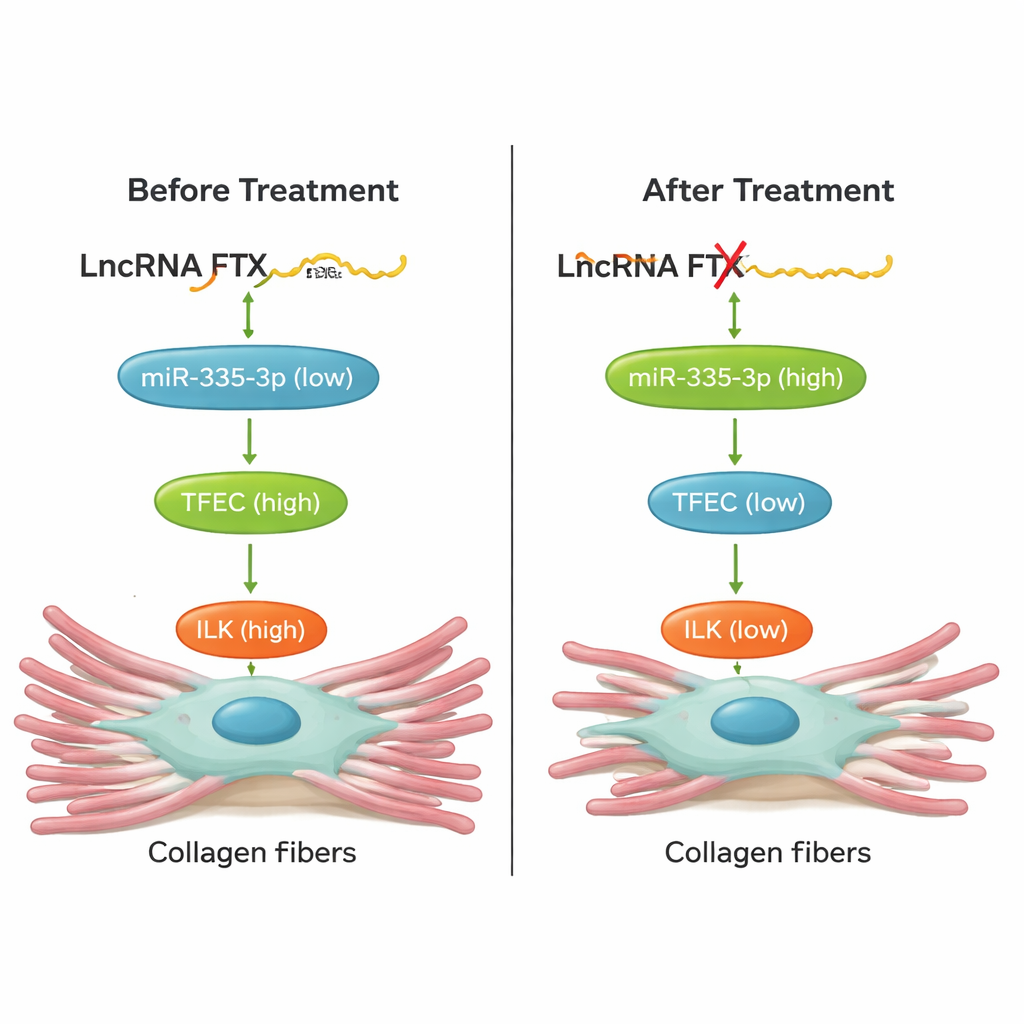
From cell culture to living hearts
Crucially, the team tested whether disrupting this chain could actually protect hearts in animals. In mice exposed to isoproterenol, turning down TFEC, knocking down FTX in the heart with an AAV9 gene therapy vector, or boosting miR‑335‑3p with a chemically stabilized "agomir" all led to less collagen buildup and lower levels of fibrosis markers in heart tissue. These interventions also improved heart function: stroke volume and ejection fraction moved back toward normal, and harmful increases in heart rate were blunted. Rescue experiments in cells showed that changing one component of the FTX/miR‑335‑3p/TFEC/ILK axis predictably shifted the others, confirming this is a tightly connected pathway rather than a loose correlation.
What this means for future treatments
To a non-specialist, the take-home message is that the authors have identified a new "control lever" for heart scarring. A long RNA called FTX lifts the brakes (miR‑335‑3p) off a master switch (TFEC), which then turns on ILK and downstream pro-scarring signals, driving excess collagen deposition and stiffening of the heart. By cutting FTX down to size, restoring miR‑335‑3p, or directly blocking TFEC, it was possible in mice to reduce scarring and improve pumping function. While more work is needed to confirm this pathway in human patients and to develop safe therapies, this RNA-based regulatory chain offers several promising points to intervene in fibrosis-driven heart failure.
Citation: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Keywords: myocardial fibrosis, heart failure, noncoding RNA, cardiac fibroblasts, fibrosis signaling