Clear Sky Science · en
Optimization of galectin-3 binding agents by in situ multiple compound synthesis and native mass spectrometry
Why this matters for future medicines
Many modern medicines work by latching onto proteins inside our bodies, but finding a small molecule that sticks tightly and selectively to just the right spot is slow, costly, and often frustrating. This study introduces a faster way to fine‑tune such molecules directly in the presence of the target protein, and then read out the winners with a highly sensitive weighing technique. The authors showcase their approach on galectin‑3, a protein linked to cancer growth, and end up with a promising drug‑like candidate that binds as strongly as some of the best existing compounds, but in an unexpected pocket on the protein surface.

Rethinking how we hunt for better drug leads
Traditional drug optimization resembles an expensive guessing game. Chemists alter a starting compound step by step, test each version, and hope to improve how tightly it binds to the target protein. But protein surfaces are flexible, water molecules get in the way, and the binding event itself can reshape the protein, making computer predictions unreliable. Even when a high‑resolution structure is available, there is no guarantee that a suggested change will help. Existing "target‑guided" methods try to let the protein choose its own partners from a pool of building blocks, yet these approaches still depend on complex analysis and indirect signals to infer which compound truly binds best.
Let the protein choose, then weigh the winners
The researchers combined two ideas into one streamlined workflow. First, they used a reversible chemical reaction that links a common sugar‑based core to many different side pieces in a single tube, forming a mixture of related molecules. By carefully adjusting the starting ratios, the resulting products settle into a balanced state governed by simple concentration rules, which helps equalize their amounts despite differences in raw reactivity. Second, they exposed this mixture to galectin‑3 and examined it with native mass spectrometry, a form of mass spectrometry that keeps protein–molecule pairs intact in a gentle, water‑like solution. Because each candidate has a distinct mass, the instrument can directly detect which molecules are actually bound to the protein, without any labels or reference markers.
From crowded mixtures to a standout binder
Using this setup, the team created dozens of galectin‑3 binders by attaching various side groups to a sugar core inspired by a known inhibitor, GB1107. They split 35 different hydrazide pieces into manageable groups, formed all the combinations in situ, and then added galectin‑3. Native mass spectrometry highlighted those compounds that most often traveled together with the protein, flagging them as primary hits. A follow‑up thermal stability test, which measures how a compound stabilizes the protein when heated, filtered out false positives caused by quirks of the gas‑phase measurement. Three leading candidates remained, and detailed heat‑based binding measurements showed that one, called GalAldBZ20, bound galectin‑3 especially tightly, in the sub‑micromolar range.
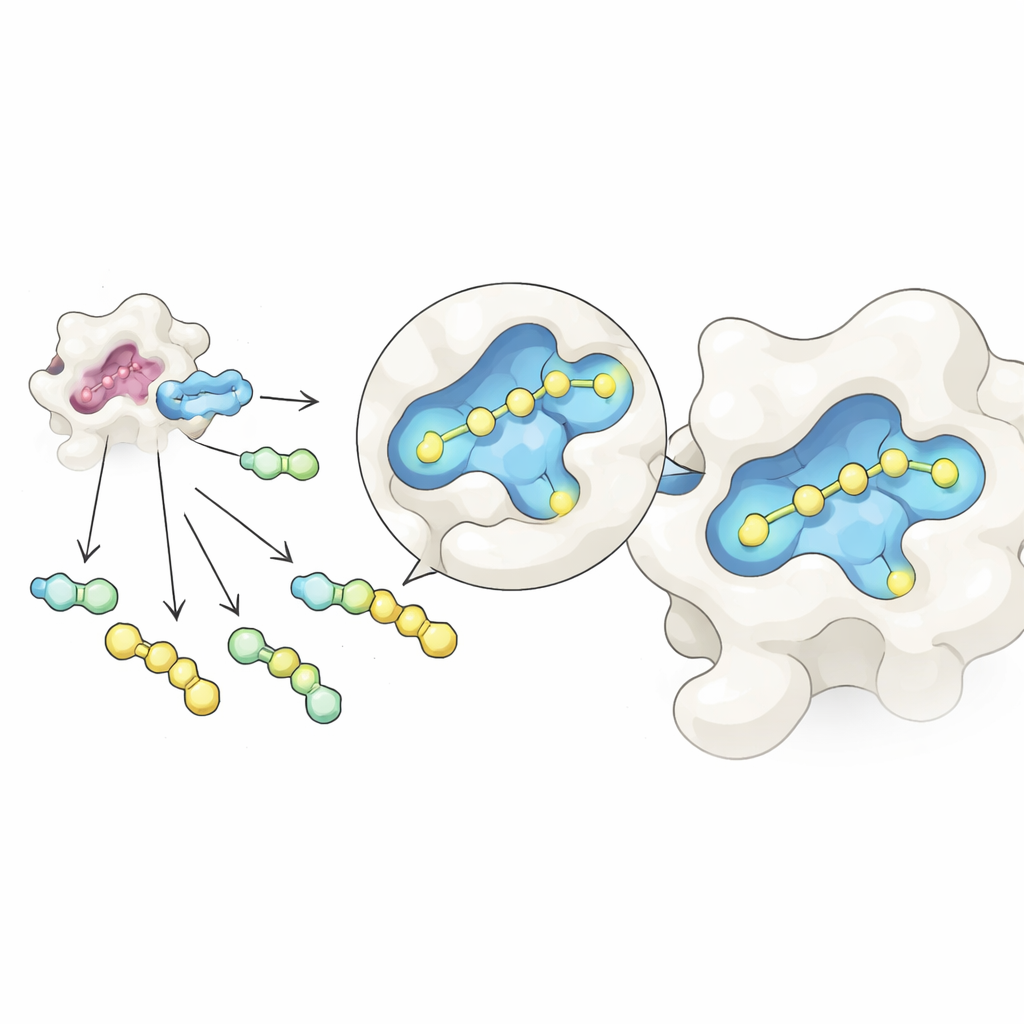
Discovering a hidden pocket and making it stronger
The next surprise came when the team looked at how GalAldBZ20 sat on the surface of galectin‑3. Most known binders use an "alpha" pocket near the sugar‑binding site, but structural methods and computer simulations indicated that GalAldBZ20 instead favored a neighboring "beta" pocket. X‑ray crystallography hinted at this, nuclear magnetic resonance in solution revealed multiple local conformations near that pocket, and molecular dynamics simulations supported a model where a nitro‑bearing ring on the molecule nestles into the beta site. Reasoning that they could lock this arrangement in more firmly, the chemists redesigned the chemical linker between the sugar and the nitro ring to encourage new polar contacts with the protein and reduce flexibility.
Turning a clever screen into a powerful candidate
With this insight, the team synthesized a small set of more rigid follow‑up molecules that kept the same sugar and nitro ring but changed the connector between them. One version, an N‑galactoside (compound 5), stood out: it bound galectin‑3 about ten times more tightly than the original hit, reaching a binding strength comparable to GB1107, yet still preferred the beta pocket. An ultra‑high‑resolution crystal structure showed clear density for the nitro ring nestled in that pocket, supported by several hydrogen bonds and a cation‑π contact with key amino acids. When the nitro group was removed or replaced with a simple methyl group, binding weakened markedly, underscoring its importance. Because galectin‑1, a related protein, lacks this beta pocket, the new compound may ultimately offer better selectivity, a prized feature in drug design.
What this means for future drug discovery
In accessible terms, this work shows that you can mix many related molecules together, let a disease‑relevant protein "choose" its favorites, and then directly weigh those protein–molecule pairs to see which stick best. Applied to galectin‑3, this strategy unexpectedly found and then strengthened binding to a less‑explored pocket, yielding a compound that rivals some of the best existing inhibitors and may serve as a lead for new anticancer drugs. More broadly, coupling in situ chemistry with native mass spectrometry offers a general shortcut for refining drug leads against proteins with multiple possible binding sites, potentially saving time, materials, and effort in the early stages of drug discovery.
Citation: Hoshi, K., Konuma, T., Taguchi, R. et al. Optimization of galectin-3 binding agents by in situ multiple compound synthesis and native mass spectrometry. Sci Rep 16, 8453 (2026). https://doi.org/10.1038/s41598-026-38570-z
Keywords: galectin-3 inhibitors, native mass spectrometry, fragment-based drug discovery, target-guided synthesis, cancer drug leads