Clear Sky Science · en
Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families
Why this matters for families and doctors
Problems with balance, walking, and feeling in the hands and feet can be deeply disabling, especially when they begin in childhood and slowly worsen over a lifetime. For many families, particularly in regions where cousins often marry, these symptoms run through several generations without a clear explanation. This study tackles a pressing question for such families in Pakistan: can modern DNA analysis finally reveal the hidden genetic causes of their ataxia (poor balance and coordination) and peripheral neuropathy (damage to the nerves in the limbs), and help doctors offer clearer diagnoses and potential treatments?
Following inherited illness through large families
The researchers worked with seven extended Pakistani families in which several members had serious movement and nerve problems. Some people mainly had ataxia, making it difficult to walk steadily or control their speech and eye movements. Others showed classic signs of peripheral neuropathy, such as muscle wasting in the hands and feet, foot deformities, and loss of reflexes. In these families, parents were related to each other, increasing the chance that children would inherit two copies of the same rare faulty gene. Using blood samples from affected and unaffected relatives, the team performed exome sequencing—reading nearly all the protein‑coding parts of the genome—to search for harmful changes that tracked with disease across the family trees. 
Pinpointing rare faulty genes
By filtering out common and harmless DNA differences, the scientists found likely disease‑causing variants in five of the seven families. Each of these families carried their own specific genetic change, and all followed a recessive pattern: people became ill only when they inherited two faulty copies, one from each parent. In one family with adult‑onset balance problems and speech difficulties, the culprit was a rare change in a gene called MFSD8, which helps shuttle materials into cell recycling compartments called lysosomes. In another family, a damaging change in AFG3L2, a protein that maintains the health of mitochondria—the cell’s power plants—was linked to childhood‑onset spastic ataxia with dystonia (abnormal muscle contractions). A third family carried a frameshift error in SETX, a gene that protects DNA during repair and is already known to cause ataxia with oculomotor apraxia, a disorder that also affects eye movements.
A closer look at inherited nerve damage
Two further families had a form of Charcot‑Marie‑Tooth (CMT) disease, a group of inherited conditions that damage the long nerves to the feet and hands. In both, the researchers found harmful variants in a gene called GDAP1, which is crucial for normal mitochondrial function in nerve cells. One GDAP1 change chopped the protein short and was associated with very severe, early‑onset disease; another swapped a single building block in the protein and produced a somewhat milder course. Strikingly, the most severely affected patient in one CMT family was also homozygous for a known disease variant in a second gene, MMACHC, which is involved in vitamin B12 processing and can sometimes be treated with vitamin‑based therapies. This double hit may explain why his symptoms were worse than those of his relatives who lacked the MMACHC variant.
When the DNA search is not enough
Not every family yielded a clear genetic answer. In two of the seven families, the team could not find any single change in the exome that convincingly matched the pattern of disease. In one case, they identified a variant in the EHHADH gene that followed the inheritance pattern but is predicted to be harmless and is known to cause a different kidney‑related condition when altered. In another, two cousins with similar movement problems turned out to have different underlying causes: one boy carried a known harmful variant in ALS2, which can lead to juvenile forms of motor neuron disease, while his affected cousins did not. These unresolved cases suggest that important mutations may lie in regions of the genome that standard exome sequencing does not capture, or that more than one subtle genetic factor might be interacting.
What this means for patients and future care
Together, the results show that powerful DNA tools can uncover the specific genes behind complex nerve and balance disorders, even in settings with limited resources. For the five families with clear findings, the work turns vague labels like “ataxia” or “neuropathy” into precise diagnoses tied to particular genes, which can guide genetic counseling, inform prognosis, and in some cases highlight treatment options, such as vitamin B12–related therapies for MMACHC‑linked disease. The study also broadens scientists’ understanding of how genes like MFSD8, AFG3L2, SETX, GDAP1, MMACHC, and ALS2 shape the health of nerve cells across the brain, spinal cord, and peripheral nerves. Looking ahead, more comprehensive genome sequencing and functional studies will be needed to solve the remaining mysteries and to translate these genetic insights into earlier diagnosis and better care for affected children and adults. 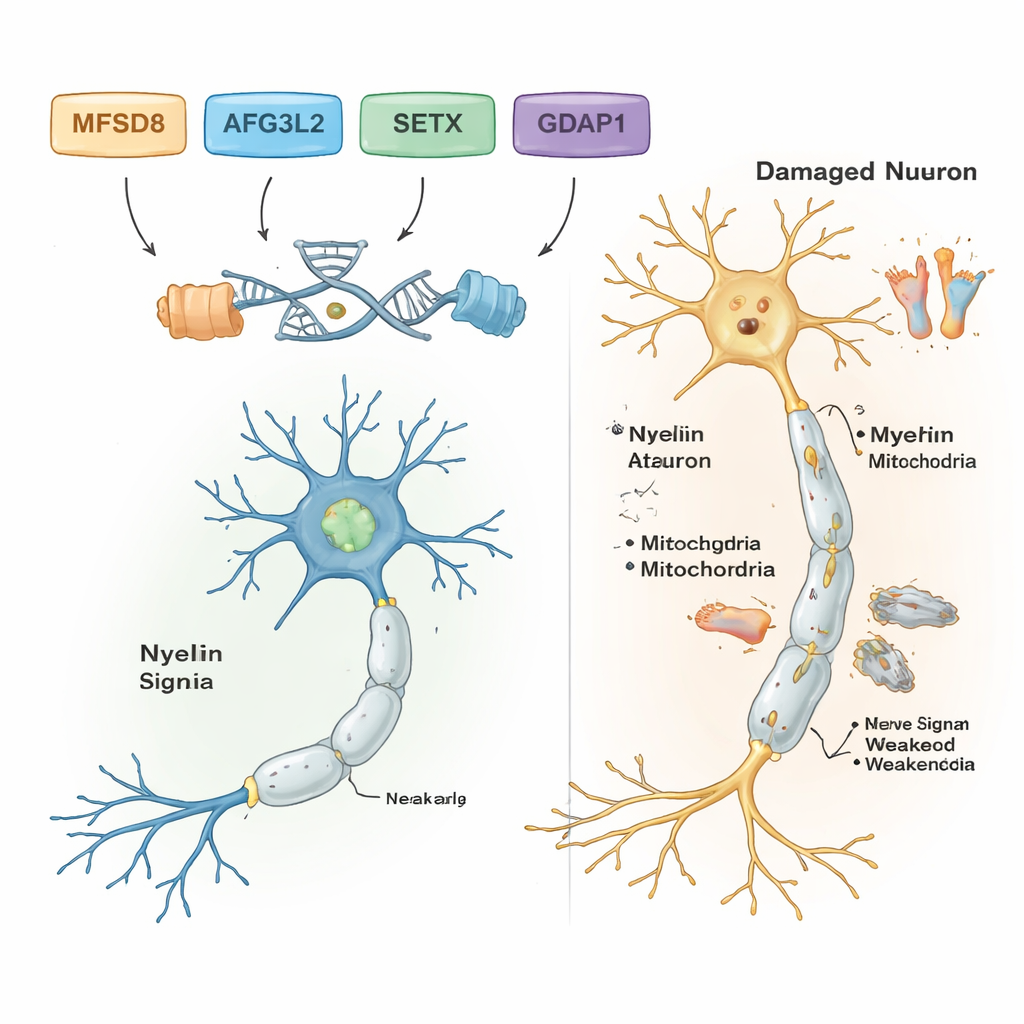
Citation: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Keywords: ataxia, peripheral neuropathy, exome sequencing, Charcot-Marie-Tooth disease, genetic diagnosis