Clear Sky Science · en
Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces
Why picking the right genes matters
Cancer tests built from modern genetic technologies can measure tens of thousands of genes at once, but doctors often have data from only a few dozen patients. Hidden in this enormous “gene jungle” are a much smaller number of signals that truly separate one cancer type from another, or a tumor from healthy tissue. This paper presents a new smart-search method for automatically picking out those key genes, aiming to make computer‑aided cancer diagnosis more accurate, faster, and easier to interpret.
Too many signals, too little data
Microarray experiments and similar technologies let researchers measure activity levels for thousands of genes in each patient sample. Yet the number of samples is usually very small, sometimes fewer than a hundred. Many of these gene readings are noisy, redundant, or irrelevant to the disease in question. Keeping all of them can overwhelm learning algorithms, slow down computation, and produce misleading models that latch onto random quirks instead of true biology. The process of trimming this down to a useful subset is called “feature selection,” and it is crucial if we want reliable predictions from high‑dimensional medical data.
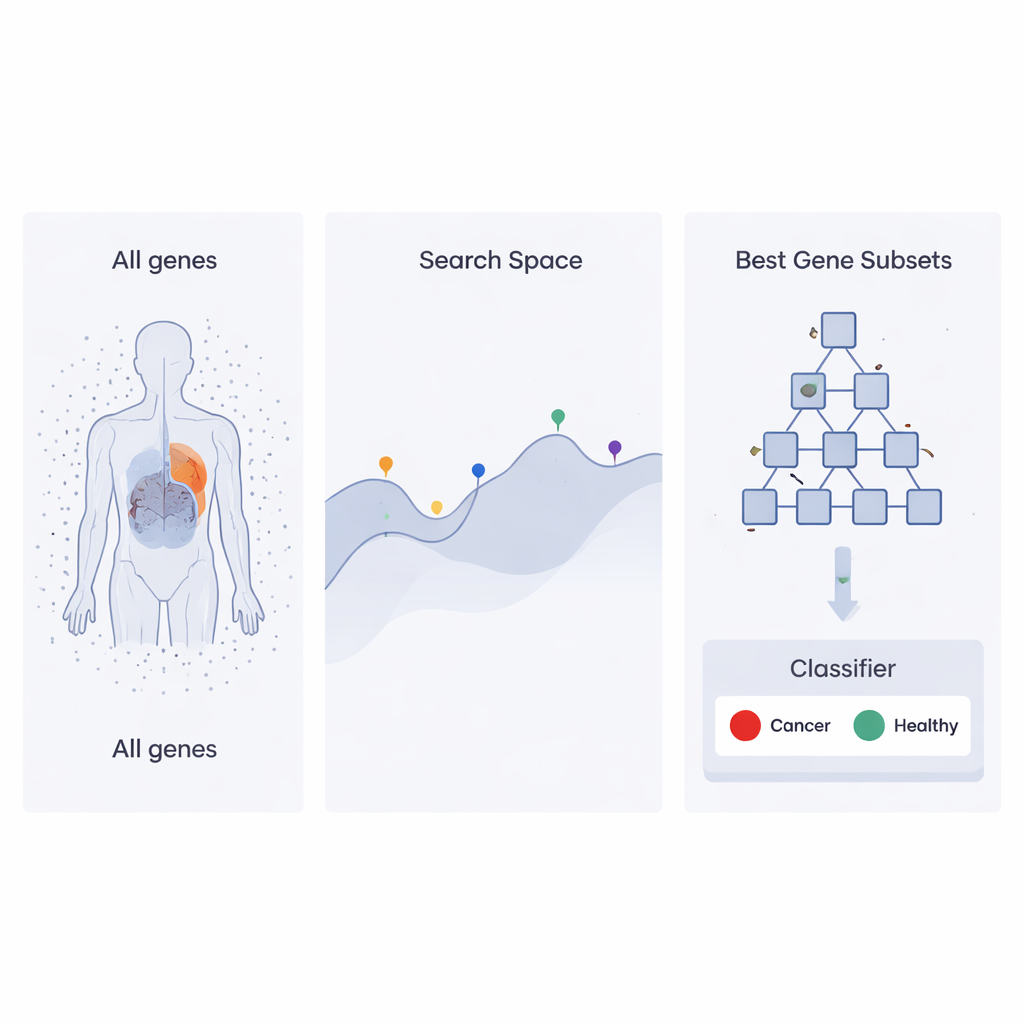
A search strategy inspired by corporate ladders
The authors build on a recent optimization approach called the Heap‑Based Optimizer (HBO), which borrows ideas from how employees are organized in a company. Imagine each possible set of genes as an “employee” whose job performance is judged by how well it helps a classifier tell cancer samples from healthy ones. These employees are arranged in a hierarchy, like a corporate ladder, using a computer structure known as a heap. High‑performing gene sets sit near the top, while weaker ones sit lower down. Over many rounds, lower‑ranked employees adjust their choices by copying and slightly modifying what their bosses and colleagues are doing, gradually pushing the whole organization toward better solutions.
Turning raw gene data into sharper patterns
To make the search more effective, the authors do not rely on raw gene readings alone. They first reshape the microarray data into an image‑like form and apply a technique called Histogram of Oriented Gradients (HOG), widely used in computer vision. HOG captures how expression levels change across genes, highlighting local patterns rather than isolated measurements. These pattern‑based features are then combined with the original gene information. A simple classifier called k‑Nearest Neighbors (KNN) serves as the “judge,” scoring each candidate gene subset by how accurately it labels new samples while also rewarding smaller, more compact sets.
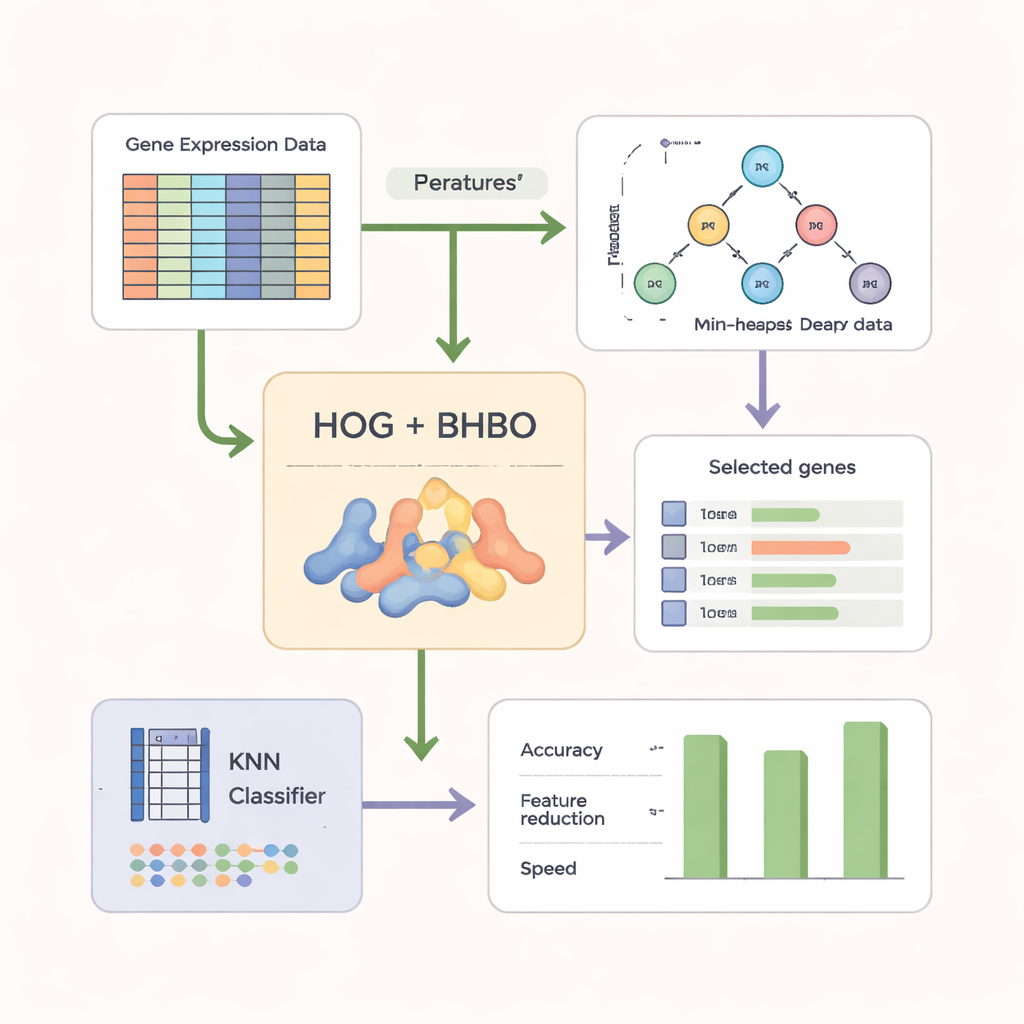
Testing on multiple cancer datasets
The researchers evaluated their binary version of the Heap‑Based Optimizer (BHBO) on nine public cancer microarray datasets, including brain tumors, leukemias, prostate cancer, and mixed tumor collections with many subtypes. Each dataset had thousands to over fifteen thousand measured genes but relatively few patient samples. For each dataset, BHBO was run repeatedly and compared with seven well‑known search methods, such as genetic algorithms and particle swarm optimization. The team measured not only accuracy, but also how many genes were kept, how quickly the search converged, and how stable the results were when the data were disturbed by simulated noise, batch effects, and label mistakes.
What the new method achieved
Across the nine datasets, the heap‑driven approach reached an average classification accuracy of about 95 percent while reducing the number of genes by more than 85 percent. It clearly beat competing methods on several datasets and showed faster convergence, meaning it homed in on good gene sets within fewer search steps. Even when the authors deliberately corrupted the data—by adding noise or flipping some sample labels—the method’s performance dropped only slightly and remained better than the alternatives. Statistical tests confirmed that these gains were unlikely to be due to chance.
What this means for future cancer diagnostics
In practical terms, this work shows that a carefully designed search strategy can sift through enormous genetic datasets and uncover small, information‑rich panels of genes that still classify cancers very well. For clinicians and researchers, such compact gene sets are easier to validate biologically, cheaper to measure in follow‑up tests, and more suitable for integration into decision‑support tools. While the method does not directly discover new drugs or pathways, it sharpens the spotlight on promising genetic markers, helping other studies focus on the most informative signals hidden in high‑dimensional cancer data.
Citation: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Keywords: cancer microarray, feature selection, metaheuristic optimization, gene biomarkers, medical data mining