Clear Sky Science · en
Computational strategies for unraveling insights from known inhibitors for further lead optimization: A case study on Celecoxib analogues
Why tiny changes in pain pills matter
Modern painkillers don’t just dull aches; they tweak the body’s chemistry in very precise ways. Celecoxib, a popular anti-inflammatory drug, targets an enzyme involved in pain and swelling while largely sparing a sister enzyme that protects the stomach. Yet dozens of close chemical cousins of celecoxib behave very differently in the body. This study uses computer modeling to ask a deceptively simple question with big implications for safer medicines: how much does a single small change in a molecule matter?
The enzyme that turns on pain
When tissue is injured or inflamed, the body releases a fatty molecule called arachidonic acid. An enzyme named COX-2 converts this molecule into prostaglandins, which trigger pain, fever and swelling. A related enzyme, COX-1, helps protect the stomach lining and platelets. Older painkillers like ibuprofen hit both enzymes, easing pain but often irritating the gut. Celecoxib was designed to slip into a slightly larger pocket found mainly in COX-2, blocking pain signals while leaving much of COX-1’s protective work intact. Understanding the detailed shape of this pocket, and how drug molecules sit inside it, is central to designing new medicines that are both powerful and safe.
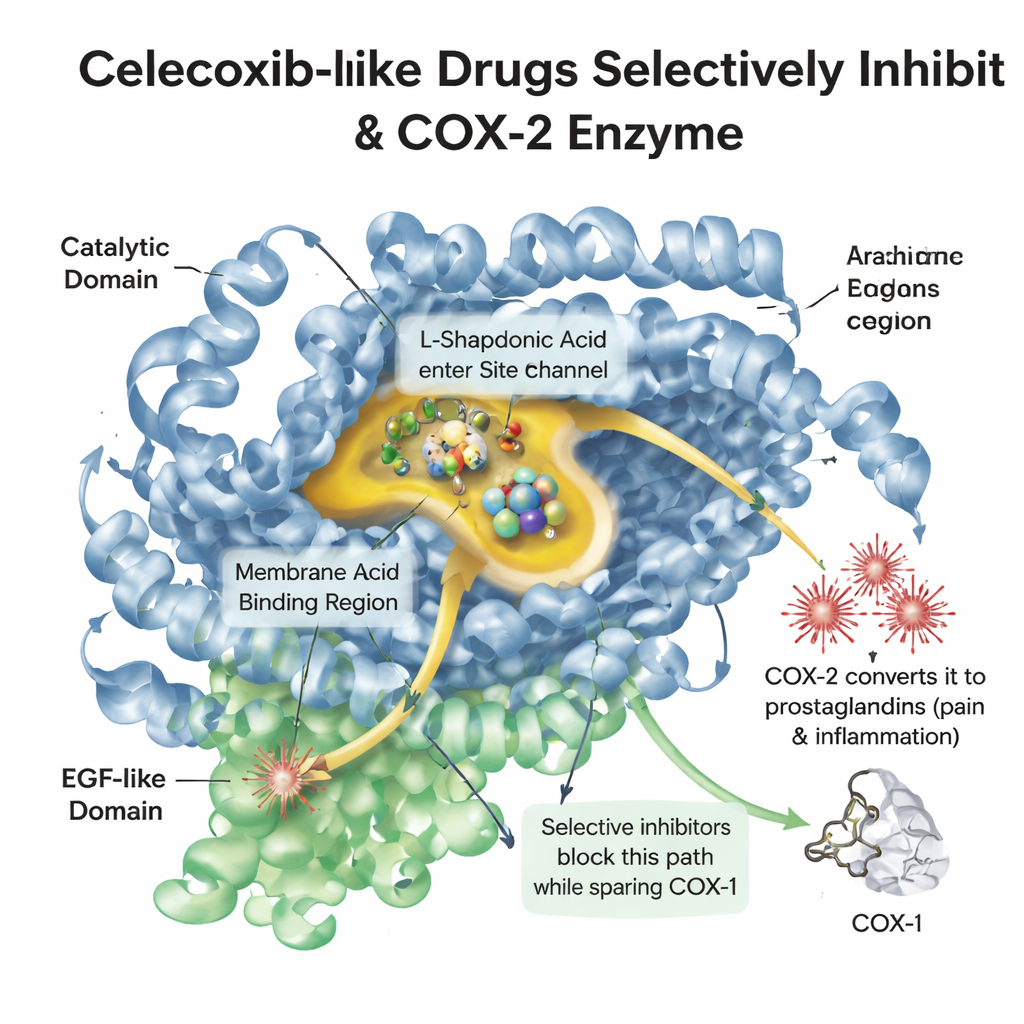
A digital library of look‑alike drugs
The researchers assembled a set of 375 molecules that all share celecoxib’s basic three-ring framework but differ in small ways, such as swapping a single atom or side group. They pulled these structures and their measured COX-2-blocking strengths from a public drug database. Using chemistry software, they generated 3D models of each molecule, calculated almost 2,000 numerical descriptors of their shapes and properties, and then docked them into a high‑resolution structure of the COX-2 enzyme. In docking, a computer nudges a molecule into the enzyme’s pocket in many ways and scores how snugly each pose fits.
What really controls strength and selectivity
The team zoomed in on three key regions of celecoxib. “Site‑1” is a ring that sits in a greasy patch of the pocket; “Site‑2” is a ring bearing a fluorine-rich tail; and “Site‑3” is a ring carrying a sulfonamide group that forms strong hydrogen bonds. Their analysis showed that Site‑1 prefers small, non‑polar groups that preserve hydrophobic contacts; making this region more water-loving, for example by adding an –OH or acid group, usually weakened the drug. At Site‑2, small electronegative groups such as fluorine often sharpened potency by improving interactions in a tight pocket, while bulkier or more polar tails tended to hurt activity. At Site‑3, the sulfonamide’s nitrogen, able to donate a hydrogen bond, was crucial; replacing it with a non‑bonding version noticeably reduced binding.
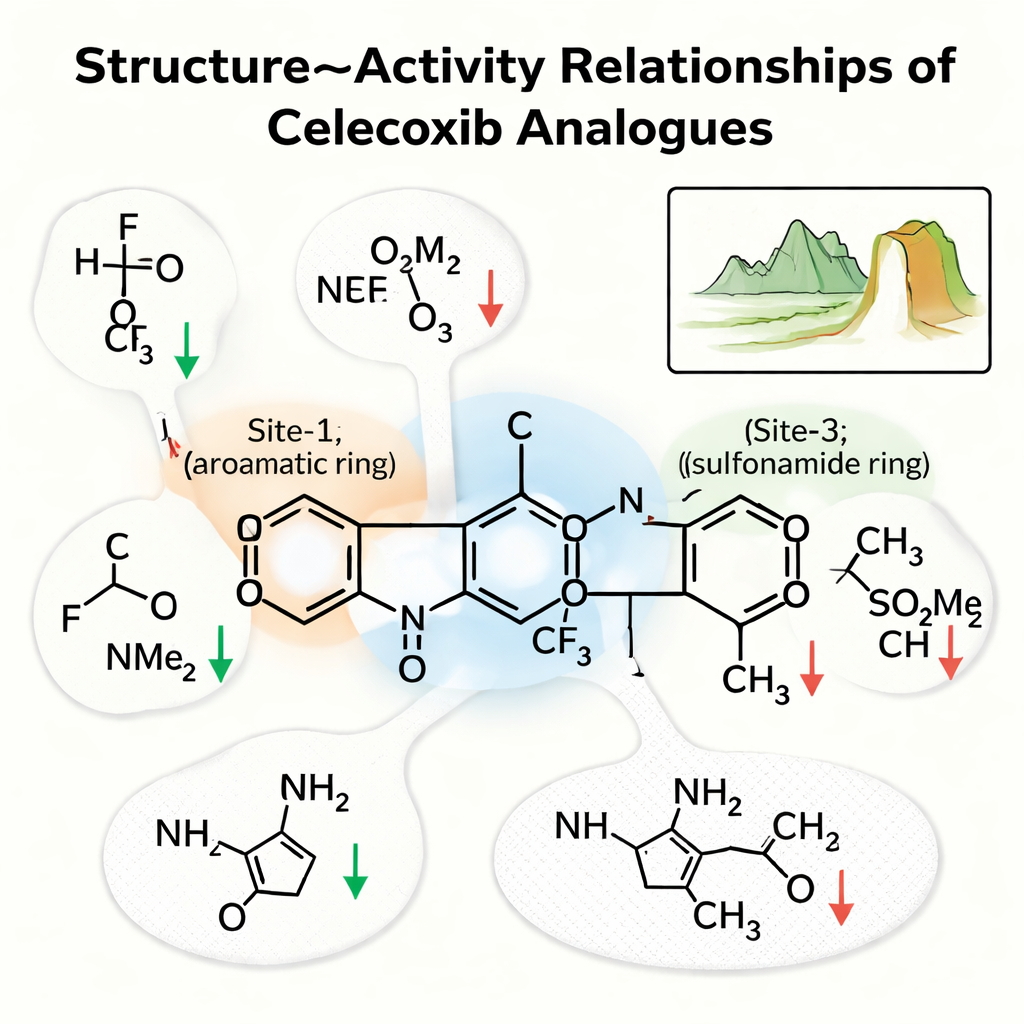
Cliffs in the chemical landscape
To move beyond simple trends, the authors built a “structure–activity landscape,” which asks how much the drug’s strength jumps when the structure changes only a little. In this view, most celecoxib-like compounds lie on gentle hills: tweaking them—say, by shifting a halogen or adding a small flexible group—nudges potency up or down in predictable ways. But a few pairs form sharp “activity cliffs,” where a tiny change, such as swapping a methyl group for a trifluoromethyl group or adding a single fluorine atom, causes a dramatic gain or loss of activity. The study also ran full molecular dynamics simulations—virtual movies of drug–enzyme complexes in motion—which confirmed that the best analogues sit stably in the pocket over hundreds of nanoseconds.
Guiding the next generation of safer pain relievers
For a non‑specialist, the take‑home message is that in drug design, small details matter enormously. Two compounds that look almost identical on paper can differ by a thousand‑fold in how strongly they block COX‑2, simply because one extra atom improves the fit in a microscopic pocket or disrupts a key contact. By systematically mapping which changes help or hurt at each of celecoxib’s three key sites, and by highlighting the dangerous “cliffs” where tiny tweaks have outsized effects, this computational work offers a roadmap for chemists. It points toward new anti-inflammatory drugs that keep the pain‑relieving punch of celecoxib while pushing safety and selectivity even further.
Citation: Grewal, S., Ghosh, B., Narayan, U. et al. Computational strategies for unraveling insights from known inhibitors for further lead optimization: A case study on Celecoxib analogues. Sci Rep 16, 6720 (2026). https://doi.org/10.1038/s41598-026-37798-z
Keywords: COX-2 inhibitors, celecoxib analogues, anti-inflammatory drugs, computational drug design, structure-activity relationships