Clear Sky Science · en
Synergistic targeting of the ARID2–MYC axis by pomalidomide and panobinostat overcomes intrinsic IMiD resistance in multiple myeloma
Why this research matters for patients
Multiple myeloma is a cancer of antibody‑producing cells in the bone marrow that has become more treatable but is still rarely curable. Many patients eventually stop responding to standard drugs, leaving doctors with fewer options. This study explores why combining two existing types of medicines—so‑called IMiDs like pomalidomide and drugs that affect DNA packaging called histone deacetylase (HDAC) inhibitors such as panobinostat—can work together to kill myeloma cells, even when the cancer is already resistant to IMiDs alone. Understanding this cooperation at the molecular level could guide better combination treatments and help more patients benefit from drugs we already have.
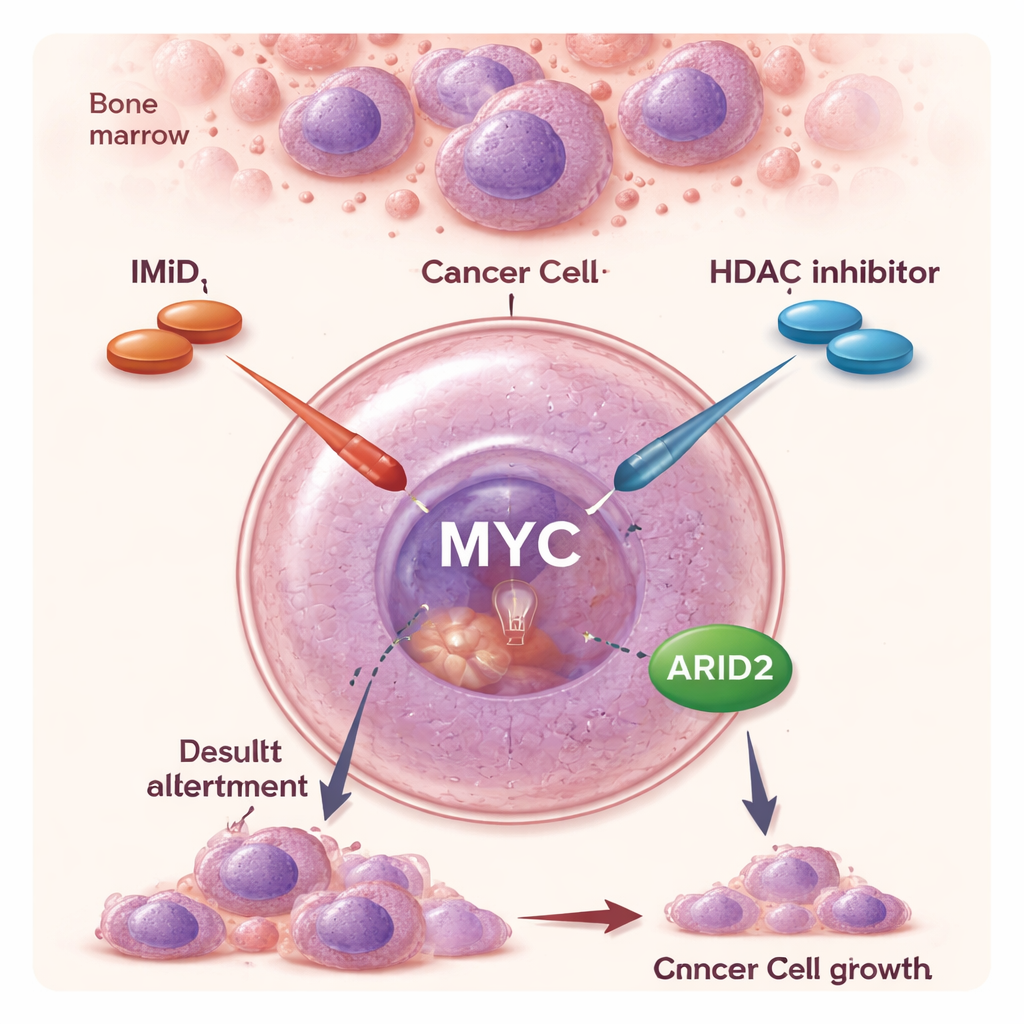
Two older drug classes, one new partnership
Over the past two decades, IMiDs and other targeted drugs have greatly extended survival for people with multiple myeloma. IMiDs act in an unusual way: instead of simply blocking a protein, they cause certain proteins to be tagged for destruction, effectively erasing them from the cell. By doing so, they weaken key survival signals that myeloma cells rely on. HDAC inhibitors such as panobinostat work differently. They loosen the tight packaging of DNA, broadly reshaping which genes are turned on or off. As single agents, HDAC inhibitors have modest effects and can cause side effects, but clinical trials hinted that pairing them with IMiDs produces a much stronger anti‑cancer response, including in patients whose disease no longer responds to IMiDs alone. The molecular reason for this synergy, however, had remained unclear.
A common pressure point: turning down MYC
The researchers systematically tested combinations of several IMiDs with different HDAC inhibitors across a panel of myeloma cell lines, using a standardized scoring system to measure how much more powerful the combinations were than each drug alone. They found that pomalidomide plus panobinostat showed particularly strong synergy in most cell models, and that this effect depended on a protein called cereblon, which IMiDs use to target their protein “victims” for destruction. By analyzing global gene activity, the team discovered that panobinostat and a related broad‑acting HDAC inhibitor strongly switched off MYC, a master growth gene often described as an “oncogene engine” in cancer cells, and that IMiDs added extra pressure on this same node. When the scientists forced myeloma cells to keep making MYC from a drug‑insensitive source, the powerful effect of the drug pair largely disappeared, showing that shutting down MYC is central to their cooperation.
Cracking resistance with an alternate route
Some myeloma cells are intrinsically resistant to IMiDs: even though the expected early targets are destroyed, MYC and other survival signals are not properly silenced, so the cells keep growing. In one such resistant model, the standard IMiD pathway linking early targets to MYC was “uncoupled.” The team asked whether an alternate route might still connect IMiDs to MYC. They focused on ARID2, a component of a large DNA‑remodeling machine called the SWI/SNF complex. Earlier work had shown that pomalidomide can mark ARID2 for destruction and that this helps lower MYC. In resistant cells, pomalidomide alone reduced ARID2 only modestly, partly because the cell increased its ARID2 production in response. When panobinostat was added, it suppressed the ARID2 gene itself, overcoming this feedback loop. Together, the two drugs strongly reduced ARID2 protein and then MYC, leading to potent cancer‑cell killing even in IMiD‑resistant lines.
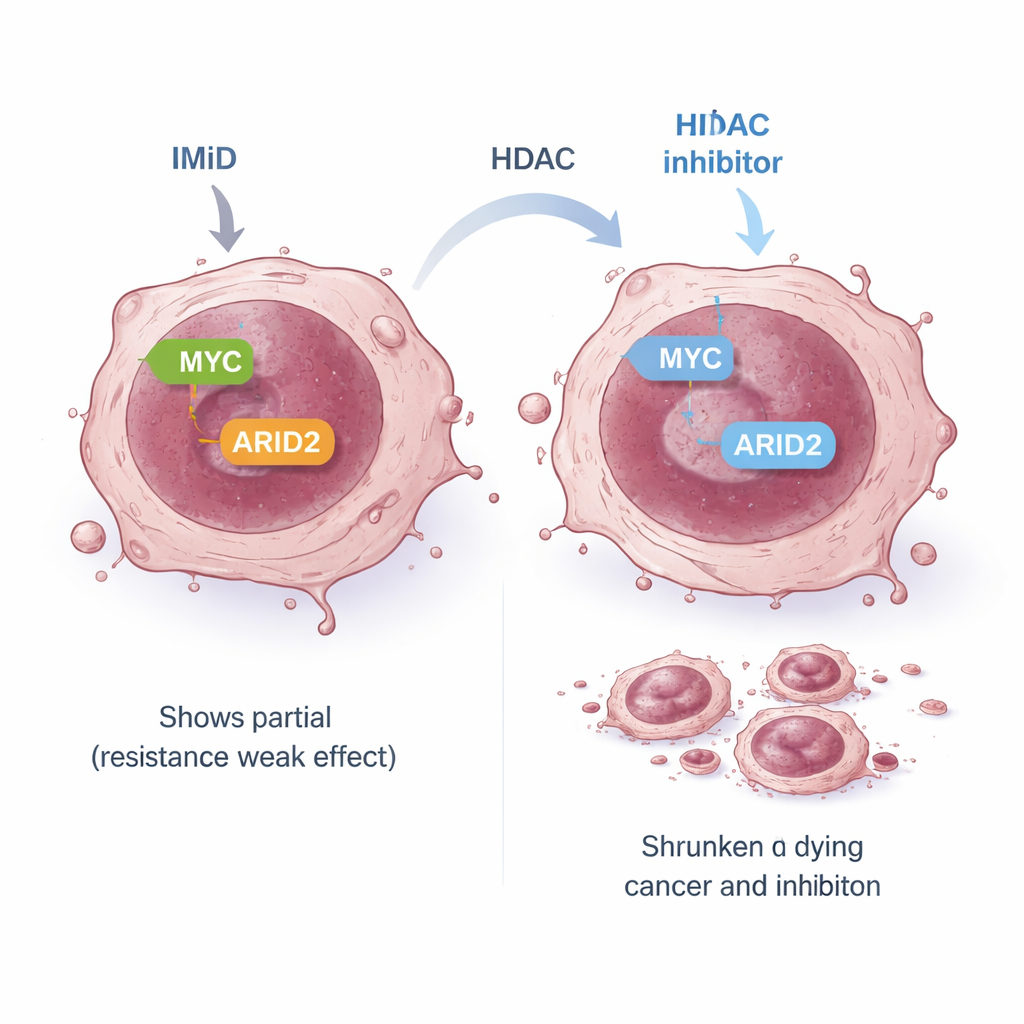
Exposing a broader weak spot in cancer cells
Because ARID2 is just one member of the SWI/SNF complex, the authors wondered whether the whole complex might be a therapeutic weak point. They found that HDAC inhibitors lower the levels of several SWI/SNF components, and that a separate small‑molecule drug designed to block the complex’s core engines (BRG1/BRM) alone can slow myeloma cell growth and reduce MYC. Importantly, this SWI/SNF inhibitor also worked synergistically with both pomalidomide and panobinostat, and a three‑drug combination pushed ARID2 and MYC down even further while strongly suppressing cell proliferation. By probing which specific HDAC enzymes were involved, the researchers highlighted HDAC1 as a key player that helps sustain the ARID2–MYC pathway, while other HDACs seem to influence MYC through parallel routes.
What this means for future myeloma treatment
For non‑specialists, the central message is that myeloma cells appear to depend on a shared “growth control hub” centered on MYC, and that there is more than one road leading into that hub. Standard IMiD therapy mainly hits one road; in some resistant cancers, that road is blocked, so MYC stays active. This study shows that an alternative road—running through ARID2 and the SWI/SNF complex—remains open, and that using panobinostat alongside pomalidomide can close it. By deliberately combining drugs that push on MYC from multiple directions, clinicians may be able to overcome some forms of built‑in drug resistance while potentially using lower doses of each medicine. Although further preclinical and clinical work is needed, the findings provide a clearer blueprint for designing smarter, mechanism‑based combinations for patients with difficult‑to‑treat multiple myeloma.
Citation: Yamamoto, J., Asatsuma-Okumura, T., Ito, T. et al. Synergistic targeting of the ARID2–MYC axis by pomalidomide and panobinostat overcomes intrinsic IMiD resistance in multiple myeloma. Sci Rep 16, 7375 (2026). https://doi.org/10.1038/s41598-026-37740-3
Keywords: multiple myeloma, drug resistance, pomalidomide, panobinostat, MYC