Clear Sky Science · en
A method for structural variant detection using Hi-C contact matrix and neural networks
Why bending DNA in 3D matters
Our DNA is usually drawn as a simple string of letters, but inside every cell it folds into a complex three-dimensional shape. When large pieces of this string are deleted, flipped, or moved around—changes called structural variations—they can disrupt genes and help drive cancer. This study introduces VarHiCNet, a new artificial intelligence system that reads 3D DNA folding maps and spots these risky large-scale changes more accurately than existing tools, offering a fresh way to study cancer genomes and other diseases.
Seeing genome changes through 3D contact maps
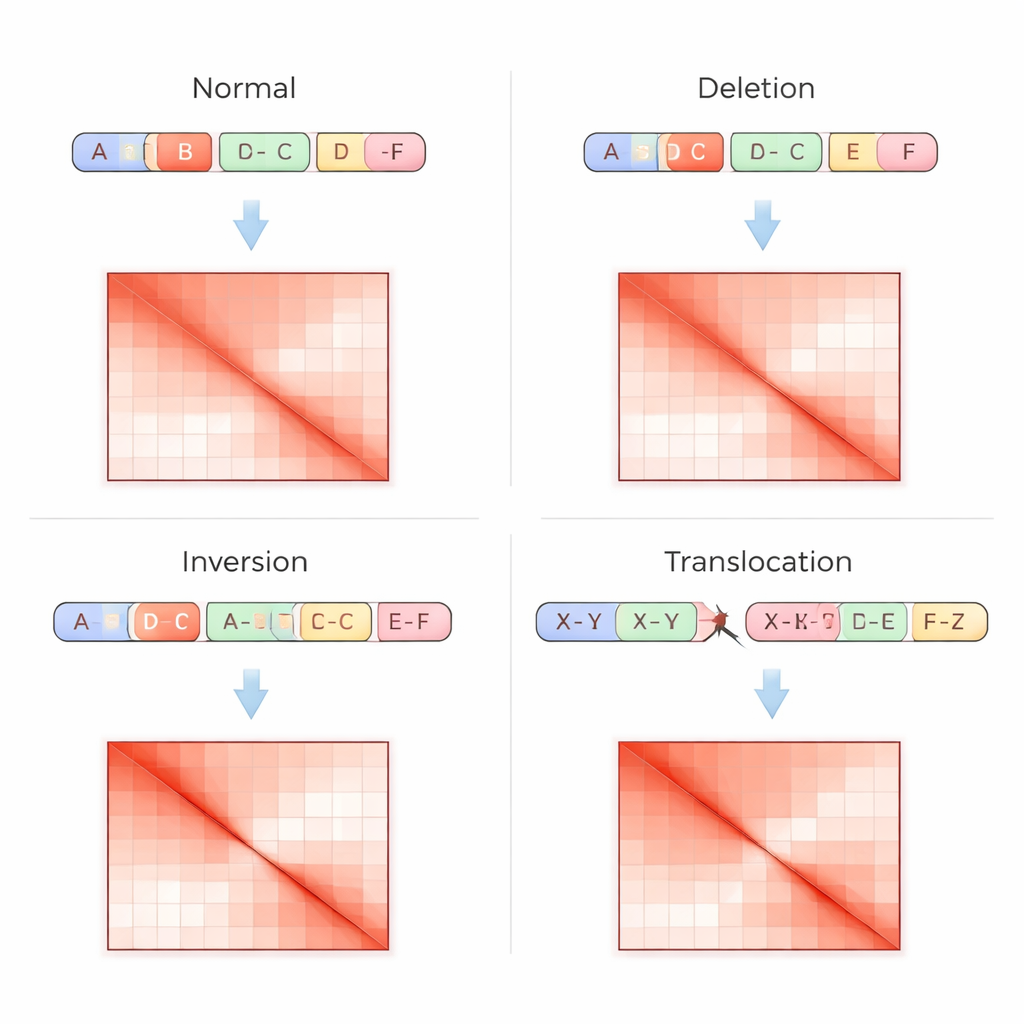
Traditional genome tests read DNA as a straight sequence, which makes it hard to spot complex rearrangements, especially in repetitive regions or when pieces are moved without changing their copy number. The Hi-C technique approaches the problem differently: it measures how often distant parts of the DNA physically touch inside the nucleus, then records these contacts as a grid, or contact matrix, where brighter spots mean stronger interaction. Structural variations leave distinctive fingerprints in these matrices—such as missing stripes where a region has been deleted, mirrored patterns when a segment is flipped, or off-diagonal hotspots where two chromosomes have been fused. VarHiCNet is designed to recognize these visual patterns automatically.
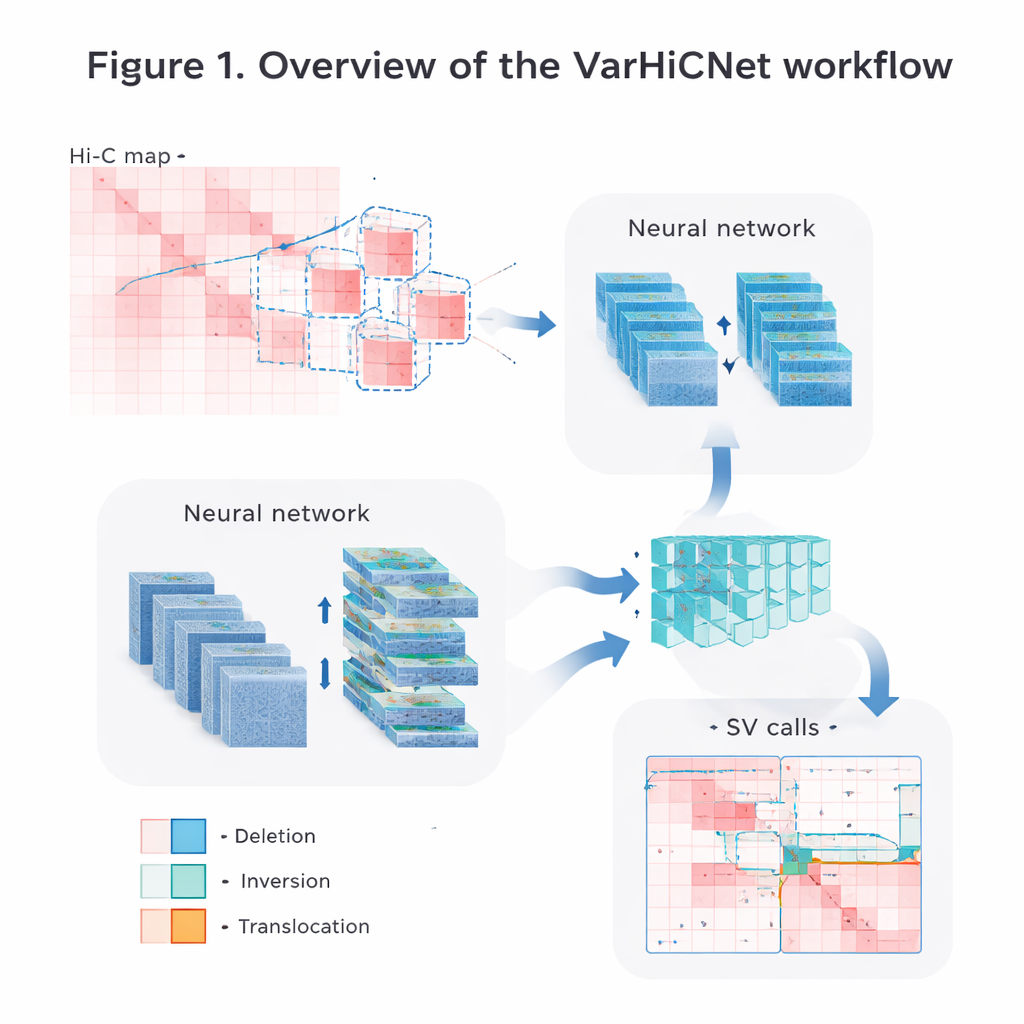
Turning genome maps into pictures for AI
The authors convert the raw Hi-C contact data into images that computer vision systems can easily process. First, they carefully normalize the matrices to correct for the natural drop in contact frequency as DNA segments get farther apart, while preserving both nearby and long-range interaction signals. Then they scan each chromosome with overlapping square windows and cut out many smaller submatrices. Each submatrix is resized into a standardized 800-by-800-pixel color image, where different contact strengths are mapped into red-toned intensities across three color channels. This image-like representation allows the model to reuse powerful techniques originally developed for recognizing objects in photographs.
Borrowing tricks from object detection
VarHiCNet treats each potential structural variant as if it were an “object” in an image. It builds on a modern object-detection framework called RT-DETR, which uses a combination of convolutional neural networks and Transformers to highlight important regions. A ResNet backbone first extracts multi-scale features: shallow layers keep fine detail needed to pinpoint exact breakpoints, while deeper layers capture broader patterns that signal large events. A feature-fusion module then blends information from several layers so that both local and global clues are preserved. Another custom block, inspired by spatial pyramid pooling, adjusts how much of the surrounding region the model “sees” at once, making it sensitive to variants that span anything from a relatively small to a very large stretch of DNA.
From candidate regions to precise variant types
Once VarHiCNet has proposed candidate regions in the Hi-C image, it must refine them into exact breakpoints and specific variant types, such as deletions, inversions, duplications, or translocations. To do this, the system zooms in on the neighborhood around each predicted breakpoint and reduces its complexity using a mathematical technique called principal component analysis, which highlights where the contact pattern changes most sharply. These compact representations are then fed into a Transformer-based classifier that learns subtle differences in the local patterns for each variant category. The result is a detailed call for each event: where it happens in the genome and what kind of structural change it represents.
Performance across diverse cancer cell lines
The researchers tested VarHiCNet on Hi-C data from six different human cancer cell lines, covering blood, breast, brain, kidney, lung, and prostate tumors. Using a high-confidence catalog of known structural variants as a gold standard, they compared their method against several leading tools that also analyze Hi-C data. Across both within-chromosome and between-chromosome events, VarHiCNet generally achieved higher or comparable F1-scores, meaning it balanced sensitivity and accuracy better than other approaches. It was particularly strong at detecting balanced translocations and inversions—rearrangements that often leave little trace in standard DNA sequencing but leave clear 3D folding signatures. The authors also showed that their design choices, such as the image resolution and feature-fusion modules, consistently improved performance in controlled tests.
What this means for understanding disease
In everyday terms, VarHiCNet gives scientists a smarter way to “look” at how the genome folds in 3D and to spot large, disease-related rearrangements that might be missed by conventional sequencing alone. By turning complex contact maps into images and applying modern vision-style neural networks, the method can detect and categorize many kinds of structural variations with high reliability across different cancer cell types. While it still struggles with some very small or highly tangled changes and depends on rich training data, VarHiCNet points toward a future in which 3D genome architecture becomes a routine part of how we read, interpret, and eventually target the genetic changes that underlie cancer and other illnesses.
Citation: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Keywords: structural variation, Hi-C, deep learning, cancer genomics, 3D genome