Clear Sky Science · en
APOL1 plasma membrane pools resist rapid protein degradation
Why a kidney protein’s “disappearing act” matters
A large share of severe kidney disease in people of recent African ancestry has been traced to two variants of a single gene, APOL1. Yet scientists still struggle to explain exactly how this gene damages kidney cells without harming most carriers. This study asks a deceptively simple question with big implications: once APOL1 protein is made inside cells, how long does it stick around, and where is it most stable? The answers reveal a surprising split personality—APOL1 is quickly destroyed inside cells but remains stubbornly stable when embedded in the cell’s outer surface, a clue that could guide future therapies.
Risk gene with a double edge
The APOL1 gene helps protect humans against certain parasites, an evolutionary advantage that likely explains why its risk variants, called G1 and G2, are common in African populations. Unfortunately, people who inherit two copies of these variants face a sharply increased risk of kidney disorders grouped as APOL1-mediated kidney diseases. Earlier work showed that when APOL1 levels rise—often in response to inflammation—the protein can become toxic, especially in delicate kidney filtering cells known as podocytes. But most studies focused on what turns APOL1 on. Much less was known about how cells turn it off again, for instance by breaking the protein down.
Tracking a fragile protein inside cells
To explore APOL1’s stability, the researchers engineered human cell lines to produce fluorescently tagged versions of APOL1 and its closest relative, APOL2. This allowed them to watch how much of each protein accumulated or disappeared under different conditions using Western blotting, microscopy, and flow cytometry. They blocked the cell’s main protein-shredding machinery, the proteasome, and separately blocked new protein production. When proteasomes were inhibited, APOL1 levels rose quickly, showing it is normally broken down at a fast pace. When new protein synthesis was stopped, APOL1 levels dropped rapidly. In stark contrast, APOL2 barely changed under either treatment, revealing it as a much more stable protein. Importantly, APOL1’s high turnover was the same for the normal version (G0) and the kidney risk versions (G1 and G2), and it held across several naturally occurring APOL1 forms that differ in how they sit in membranes.
Sequence clues and a tale of two neighborhoods
Diving into the protein’s structure, the team used computer tools to scan APOL1 and APOL2 for floppy, unstructured segments known as intrinsically disordered regions. Such regions often act as “eat me” signals for the proteasome. They identified two strong candidate regions in APOL1 that were largely absent in APOL2. To test whether APOL1’s unique front end contributes to its fragility, they created hybrids: a shortened APOL1 missing its first 59 amino acids, and an APOL2 chimera carrying that APOL1 segment. Adding APOL1’s N‑terminal piece to APOL2 made APOL2 degrade more quickly, while the truncated APOL1 remained unstable, suggesting that more than one part of APOL1 encourages rapid breakdown. Together, these results link APOL1’s unusual flexible segments to its fast turnover, without tying this behavior specifically to the disease-causing variants.
Stubborn protein at the cell surface
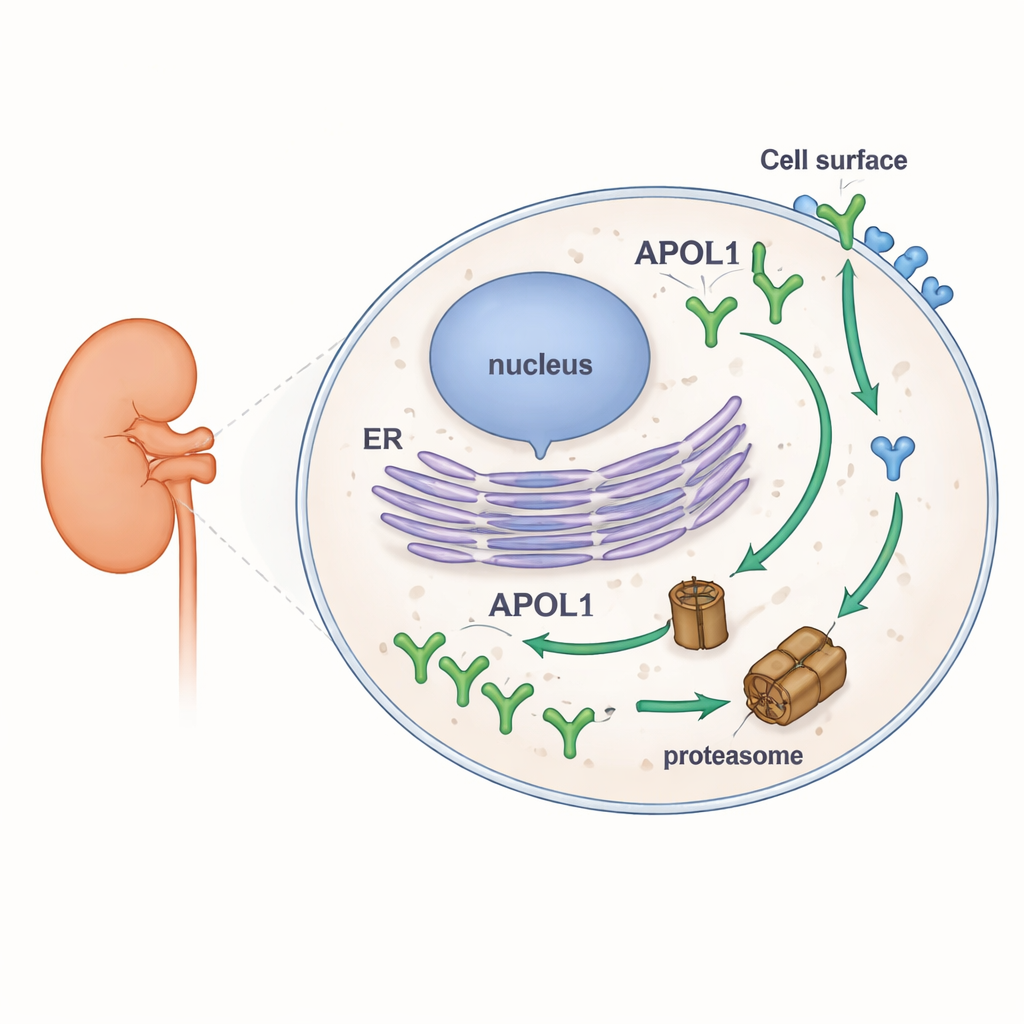
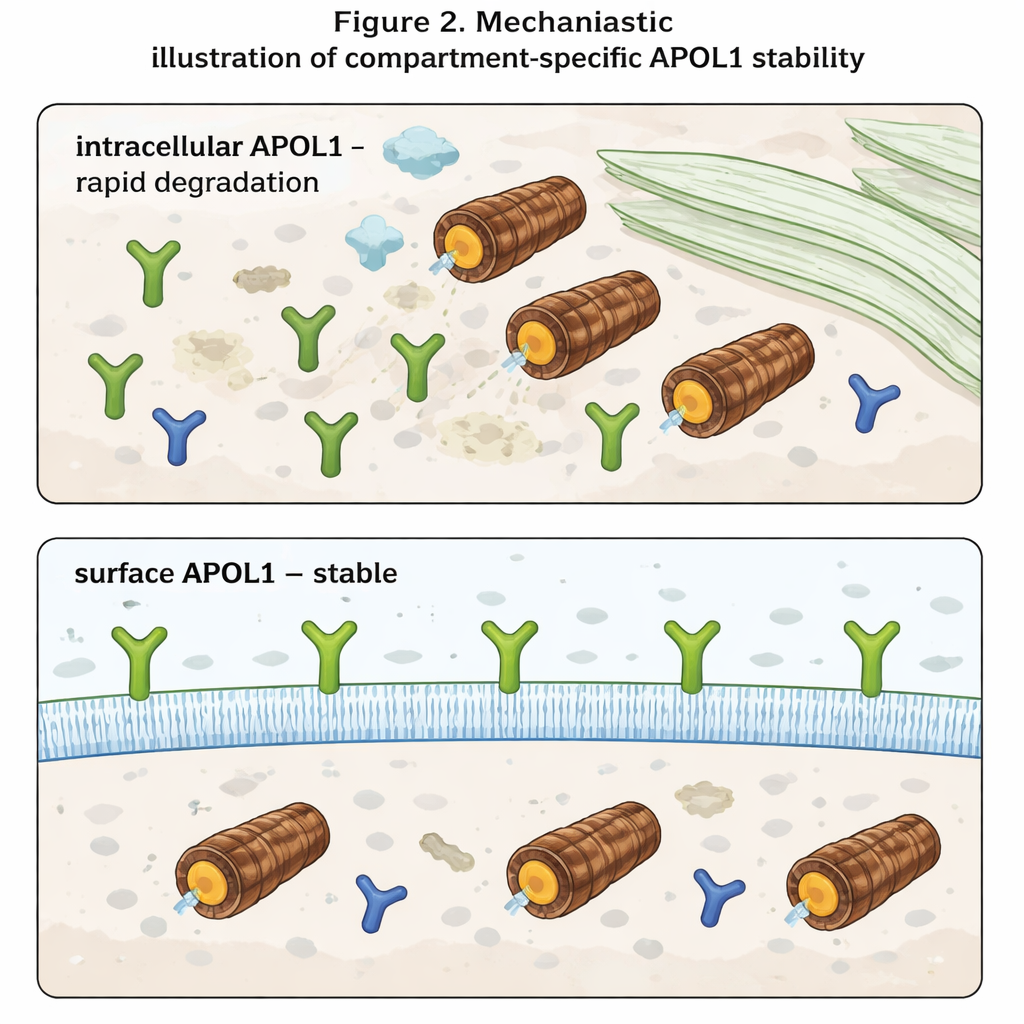
The most striking finding emerged when the authors distinguished between APOL1 inside the cell and APOL1 on the cell surface. Using antibodies that recognize only the portion of APOL1 exposed to the outside, they measured surface levels separately from total levels. Inside the cell, APOL1 behaved as expected: it piled up when proteasomes were blocked and vanished quickly when new synthesis was stopped. Surface APOL1, however, hardly budged under either condition. Once APOL1 molecules reached the plasma membrane, they proved highly resistant to rapid degradation. Moreover, even though risk variants produced less total APOL1 than the normal version, their surface levels were similar. This suggests that risk and normal APOL1 are cleared at comparable rates inside the cell, but that the membrane-embedded pools—which are thought to form ion channels and drive toxicity—are preserved in all variants.
What this means for future treatments
For non-specialists, the take-home message is that APOL1 behaves very differently depending on where it resides. Inside the cell, it is a short-lived protein, quickly recognized and destroyed. At the cell surface, it becomes long-lived and relatively protected, even when the cell’s degradation machinery is altered. Because disease seems to arise when APOL1 channels at the surface disturb the balance of ions like sodium and potassium, therapies may need to focus less on total APOL1 levels and more on how much of it reaches and persists in the plasma membrane. Strategies that reduce trafficking of APOL1 to the surface or selectively destabilize the surface pool could, in principle, blunt kidney damage without fully blocking the gene’s beneficial immune functions.
Citation: Höffken, V., Alvermann, L., Niggemeier, D. et al. APOL1 plasma membrane pools resist rapid protein degradation. Sci Rep 16, 6718 (2026). https://doi.org/10.1038/s41598-026-37647-z
Keywords: APOL1, kidney disease, protein degradation, plasma membrane, proteasome