Clear Sky Science · en
In vitro characterization of the catalytic domain of human histone deacetylase 5
Why tiny switches in our DNA packaging matter
Inside every cell, our DNA is wrapped around proteins that act like spools, helping fit meters of genetic material into a microscopic space. Whether a gene is turned on or off often depends on small chemical tags on these spool proteins. This study zooms in on one particular protein "switch" called HDAC5, which is linked to heart disease, brain disorders, cancer, and more. By understanding how HDAC5 works at the molecular level, the researchers hope to pave the way for more precise medicines with fewer side effects.
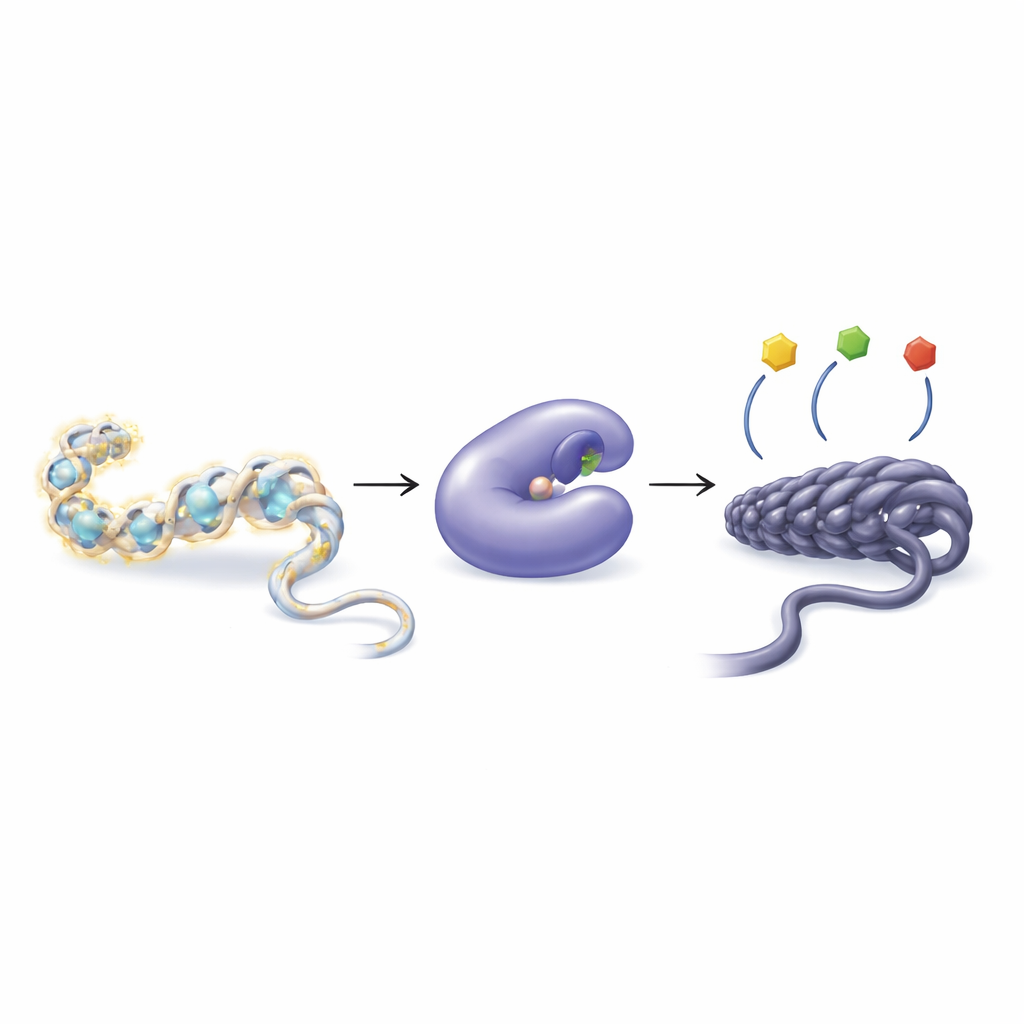
How cells tune genes with tiny chemical tags
Our DNA does not float freely but is wrapped around proteins called histones, forming a structure known as chromatin. Cells can attach or remove small chemical groups, such as acetyl groups, from histone tails to make the chromatin looser or tighter. Looser packing generally makes genes easier to read; tighter packing tends to silence them. Two groups of enzymes manage this balance: histone acetyltransferases add acetyl groups, while histone deacetylases (HDACs) remove them. When this balance is disturbed, it can contribute to a wide range of diseases including cancer, heart problems, muscle wasting, and immune disorders.
Why HDAC5 is a promising but tricky drug target
HDACs form a large family of related enzymes divided into several classes. Many of the medicines now in clinical use block many HDAC types at once, which can shut down important normal functions and cause strong side effects. Class IIa HDACs, including HDAC5, stand out because they are enriched in specific tissues like brain, heart, and skeletal muscle, and they partner with other proteins to regulate key gene networks. HDAC5 often acts as a bridge, bringing a highly active partner enzyme (HDAC3) to certain genes so that chromatin can be tightened and those genes silenced. Because of these focused roles, HDAC5 is seen as an attractive target for more selective drugs, but there has been a lack of detailed biochemical data and no high-resolution structure of its active core, making rational drug design difficult.
Rebuilding HDAC5 in a test tube
To tackle this gap, the researchers produced only the catalytic core of human HDAC5 – the part that carries out the chemical reaction – in bacteria, purified it, and confirmed that it forms a stable, single-unit protein in solution. They then tested how well it works under different salt levels and acidity. HDAC5’s activity remained robust across a wide range of salt and peaked at mildly basic conditions, similar to those inside many cells. Using special fluorescent test molecules, they found that the natural form of HDAC5 recognizes only one particular type of substrate commonly used to probe class IIa enzymes. Guided by earlier work on related HDACs, they swapped a single amino acid (histidine) for a tyrosine at a critical spot. Remarkably, this small change allowed the mutant version of HDAC5 to process both kinds of test substrates efficiently, revealing how a single residue in the active site steers the enzyme’s chemical preferences.
Testing and comparing two new drug candidates
The team next examined two experimental HDAC5-blocking molecules, known as NT160 and FFK24. These compounds use a newer zinc-binding group that avoids some of the toxicity and poor selectivity seen with older, hydroxamate-based drugs. By measuring how each inhibitor slowed HDAC5 in carefully controlled reactions, the authors determined extremely low inhibition constants in the nanomolar range, meaning both compounds stick tightly to the enzyme. NT160 consistently bound about ten times more strongly than FFK24. To understand why, the researchers used computer docking with an AlphaFold-predicted structure of HDAC5’s core. Both inhibitors shared a common head region that nestled deep into the active pocket and contacted the metal ion, but NT160’s tail formed extra stabilizing contacts with specific amino acids in the pocket. These additional interactions likely explain its greater potency.
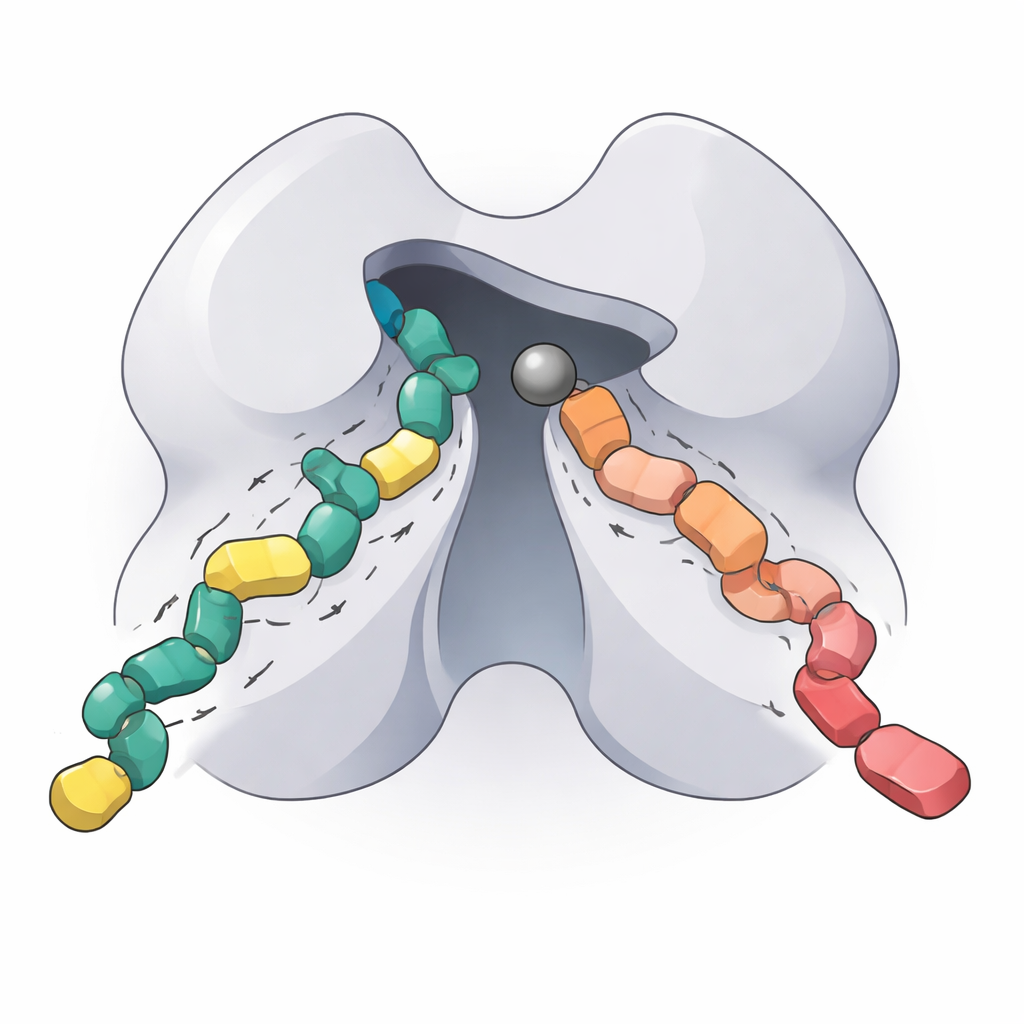
What this means for future targeted therapies
By reconstituting the working core of HDAC5, mapping its optimal working conditions, dissecting how a single amino acid change alters its behavior, and quantifying how two next-generation inhibitors bind, this study delivers a detailed biochemical "fingerprint" of an important but previously undercharacterized enzyme. For non-specialists, the key takeaway is that HDAC5 helps control whether certain genes are on or off, and that precisely tuning this switch could be valuable in treating heart disease, neurodegeneration, cancer, and immune disorders. The new insights and tools presented here should help researchers design HDAC5- and class IIa–selective drugs that act where they are needed while minimizing unwanted effects elsewhere in the body.
Citation: Mammen, C., Hornung, F.M., Anzenhofer, C. et al. In vitro characterization of the catalytic domain of human histone deacetylase 5. Sci Rep 16, 7935 (2026). https://doi.org/10.1038/s41598-026-37633-5
Keywords: histone deacetylase 5, epigenetic regulation, HDAC inhibitors, targeted cancer therapy, chromatin structure