Clear Sky Science · en
A DNABERT based deep learning framework for predicting transcription factor binding sites
Why predicting DNA control switches matters
Every cell in your body carries essentially the same DNA, yet brain cells, liver cells and immune cells behave very differently. One reason is that special proteins called transcription factors act like molecular switches, turning genes on or off by docking at short stretches of DNA known as binding sites. Experimentally finding all these docking spots across the genome is slow and expensive. This study introduces TFBS-Finder, a new artificial intelligence model that can read raw DNA letters and more accurately predict where transcription factors bind, potentially speeding up research into gene regulation and disease.
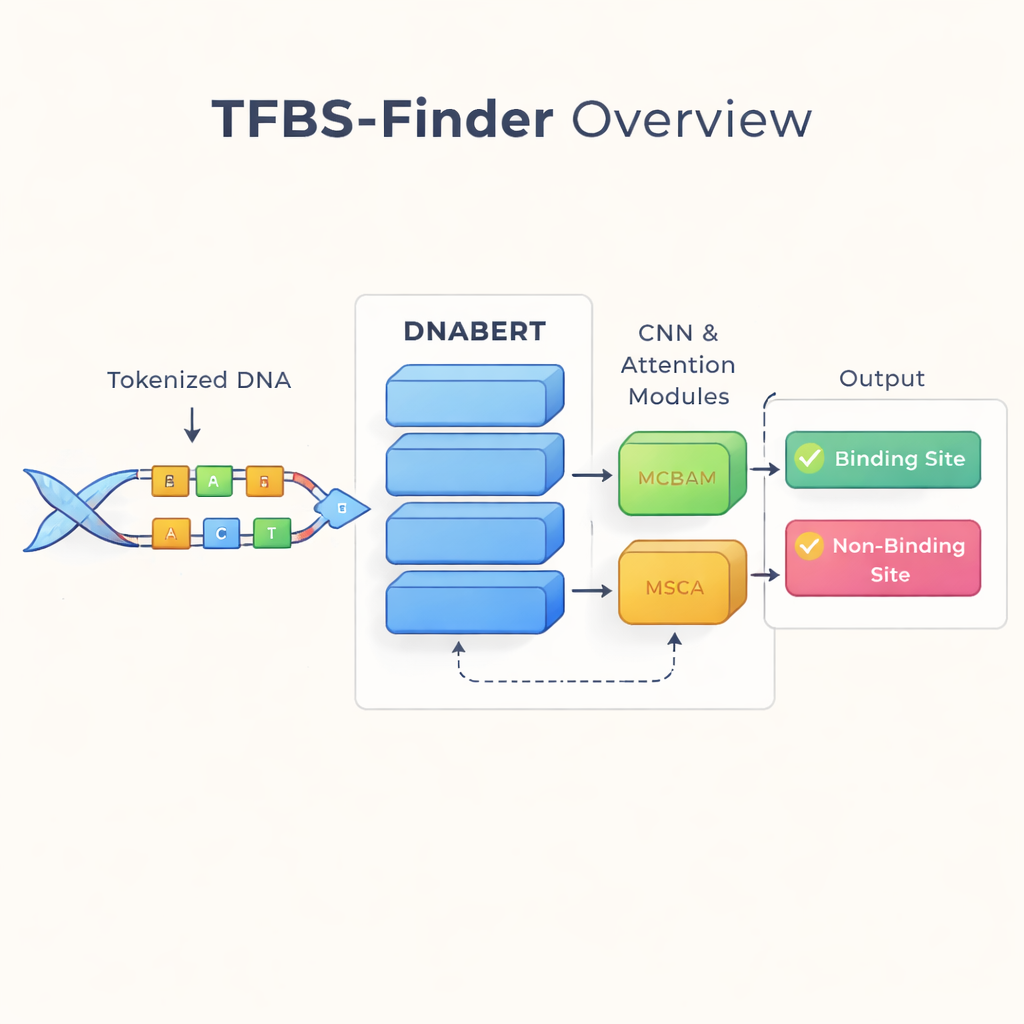
Reading DNA like a language
The authors build on an idea that has transformed language technology: treat DNA as if it were text. They use DNABERT, a version of the BERT language model retrained on human DNA instead of words. DNABERT does not look at single letters alone; it breaks DNA into overlapping short “words” of five letters and learns how these pieces tend to co-occur. This allows the model to capture long-range context, such as how patterns at one end of a sequence relate to patterns far away, much like understanding the meaning of a sentence rather than isolated words.
Finding local patterns with focused attention
While DNABERT is good at grasping global context, transcription factor binding often depends on very short, precise motifs—local patterns in the DNA. TFBS-Finder therefore adds several extra components on top of DNABERT. A convolutional neural network (CNN) combs through the sequence embeddings to highlight recurring local shapes, akin to how image software detects edges and corners. Two attention modules, called MCBAM and MSCA, then act like adjustable spotlights, strengthening the most informative features and downplaying noise. Together, these blocks balance big-picture context with fine-grained details to decide whether a DNA segment contains a true binding site.
Proving each piece really helps
To test whether all these components are necessary, the team performed extensive “ablation” experiments, systematically removing or rearranging modules and retraining the system on 165 benchmark datasets covering 29 transcription factors across 32 cell types. Using standard measures of prediction quality, the full TFBS-Finder model consistently came out on top. Simpler versions that relied only on DNABERT, or left out one of the attention modules, clearly lost accuracy. Statistical tests confirmed that these drops in performance were not due to chance, showing that the combination of global sequence understanding and carefully designed attention to local patterns is crucial.
Working across cell types and beating older tools
An important question is whether a model trained in one biological context can generalize to another. The authors focused on a well-studied transcription factor called CTCF and trained TFBS-Finder on data from one cell line, then tested it on others. In all combinations, the model reached high scores, suggesting that it captures core features of CTCF binding that are shared across tissues. When compared against nine leading methods, including earlier deep learning and BERT-based models, TFBS-Finder showed higher average accuracy and produced more reliable rankings of binding sites. It also ran slightly faster and used less memory than the most similar previous model, indicating that better performance did not require heavier computation.
Seeing what the model has learned
Complex AI systems are often criticized as “black boxes.” Here, the researchers tried to open that box by visualizing which DNA positions most influenced TFBS-Finder’s decisions. For two transcription factors with well-known binding motifs, CEBPB and GATA3, they generated importance scores along the sequence and clustered the strongest signals into consensus patterns. These recovered motifs closely matched reference motifs from established databases, and the predicted binding regions overlapped with independently detected motif instances. This suggests that TFBS-Finder is not simply memorizing examples but has learned biologically meaningful rules about how transcription factors recognize DNA.
What this means for genetics and medicine
TFBS-Finder provides a more accurate and interpretable way to map the control switches embedded in our DNA. By pinpointing where transcription factors are likely to bind, it can help researchers chart gene regulatory networks, prioritize which genetic variants might disrupt crucial control sites, and design more targeted experiments. Although the current work uses shuffled sequences as artificial negatives and focuses only on DNA letters, the authors plan to add structural information about DNA shape and explore more realistic background sequences. As these models improve, they could become powerful aids for understanding how changes in noncoding DNA contribute to development, evolution and disease risk.
Citation: Dutta, P., Ghosh, N. & Santoni, D. A DNABERT based deep learning framework for predicting transcription factor binding sites. Sci Rep 16, 7018 (2026). https://doi.org/10.1038/s41598-026-37483-1
Keywords: transcription factor binding sites, deep learning, DNABERT, gene regulation, genomics