Clear Sky Science · en
GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss
Why tiny ear cells matter for children’s futures
Hearing loss present at birth affects millions of children worldwide, often shaping how they learn to speak, succeed in school, and connect with others. One of the most common genetic culprits is a gene called GJB2, but doctors have not fully understood how changes in this gene actually damage the inner ear. This study uses zebrafish and human cells to trace the chain of events from a single DNA change in GJB2 to the death of delicate sound-sensing cells, and points to a new molecule, IFI27, as a possible target for future treatments.
A common gene change behind silent childhood
The researchers began by screening blood samples from 1,199 children with suspected hereditary hearing loss in Fujian Province, China. They focused on several well-known deafness genes and found that changes in GJB2 dominated the picture, accounting for 85% of all detected mutations. Among these, a specific change called c.109G>A (also known as p.Val37Ile) was the most frequent. This variant is relatively common in the general population yet strongly enriched in people with hearing loss, suggesting it plays a major role in non-syndromic hearing loss—hearing problems that occur without other medical issues.
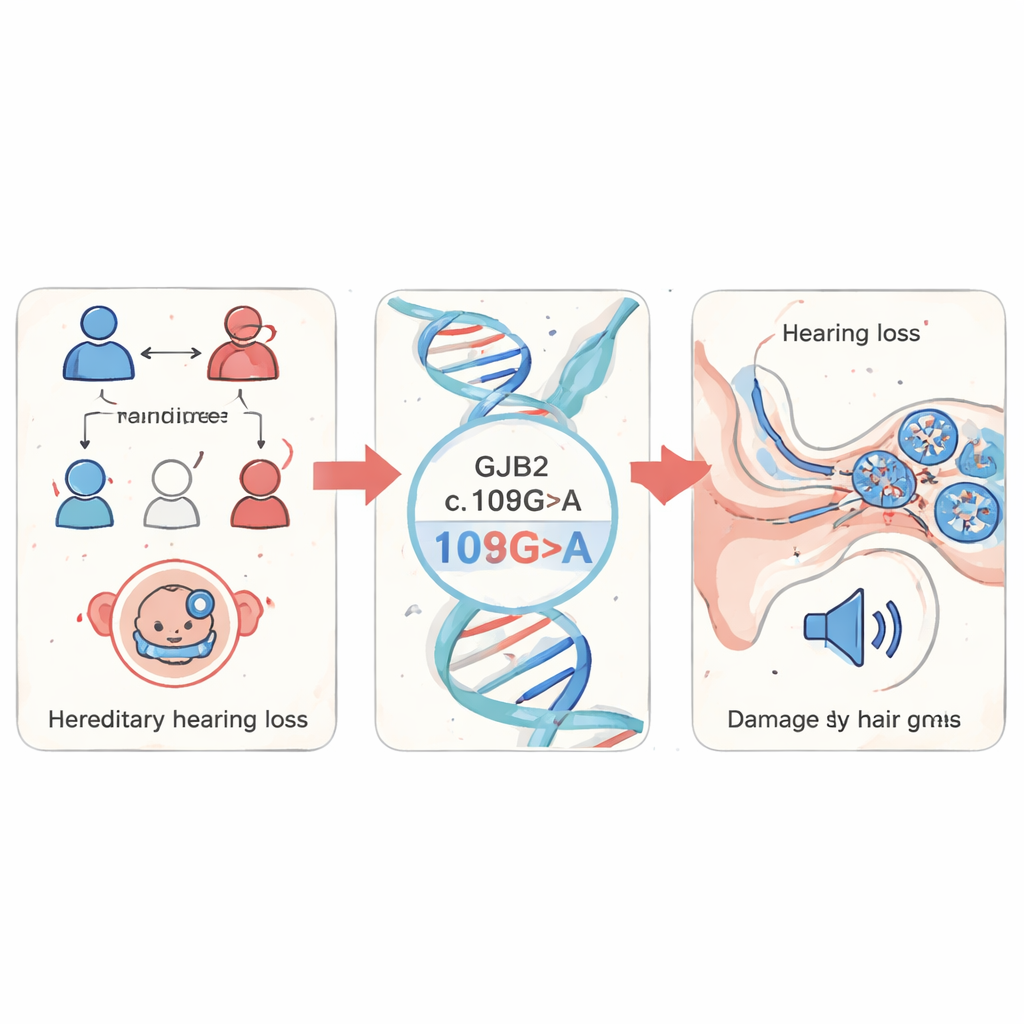
Following the damage in a transparent fish
To see what this mutation does in a living organism, the team turned to zebrafish, a small freshwater fish whose embryos are see-through and share many genes and ear structures with humans. They engineered zebrafish embryos to produce either normal human GJB2 or the c.109G>A mutant version, and also used a “knockdown” approach to reduce the fish’s own gjb2 gene. Embryos with the mutant or reduced gene showed delayed growth, curved tails, and swelling around the heart, signs that development was going off track. Most importantly, their inner ears were clearly abnormal: key structures called otoliths were smaller and farther apart, and the fluid-filled cochlear region was reduced in size. When the scientists added back normal GJB2 alongside the mutant, many of these structural problems improved, showing that the mutation itself was driving the defects.
From faulty ears to poorer hearing behavior
Because hearing depends on tiny “hair cells” that convert sound vibrations into nerve signals, the team stained these cells in the zebrafish. Fish with the GJB2 mutation or knockdown had far fewer hair cells in both the inner ear and along the body surface, where zebrafish also sense water movement. The researchers then tested how well the fish responded to sound. Using an automated tracking system, they measured how far and how fast 5-day-old larvae swam when exposed to brief bursts of sound. Normal and wild-type GJB2 fish reacted by swimming more and faster, while mutant and knockdown fish barely changed their behavior, indicating impaired hearing. Again, adding normal GJB2 partly restored both hair cell numbers and sound-driven movement.
A killing pathway inside the cell’s powerhouses
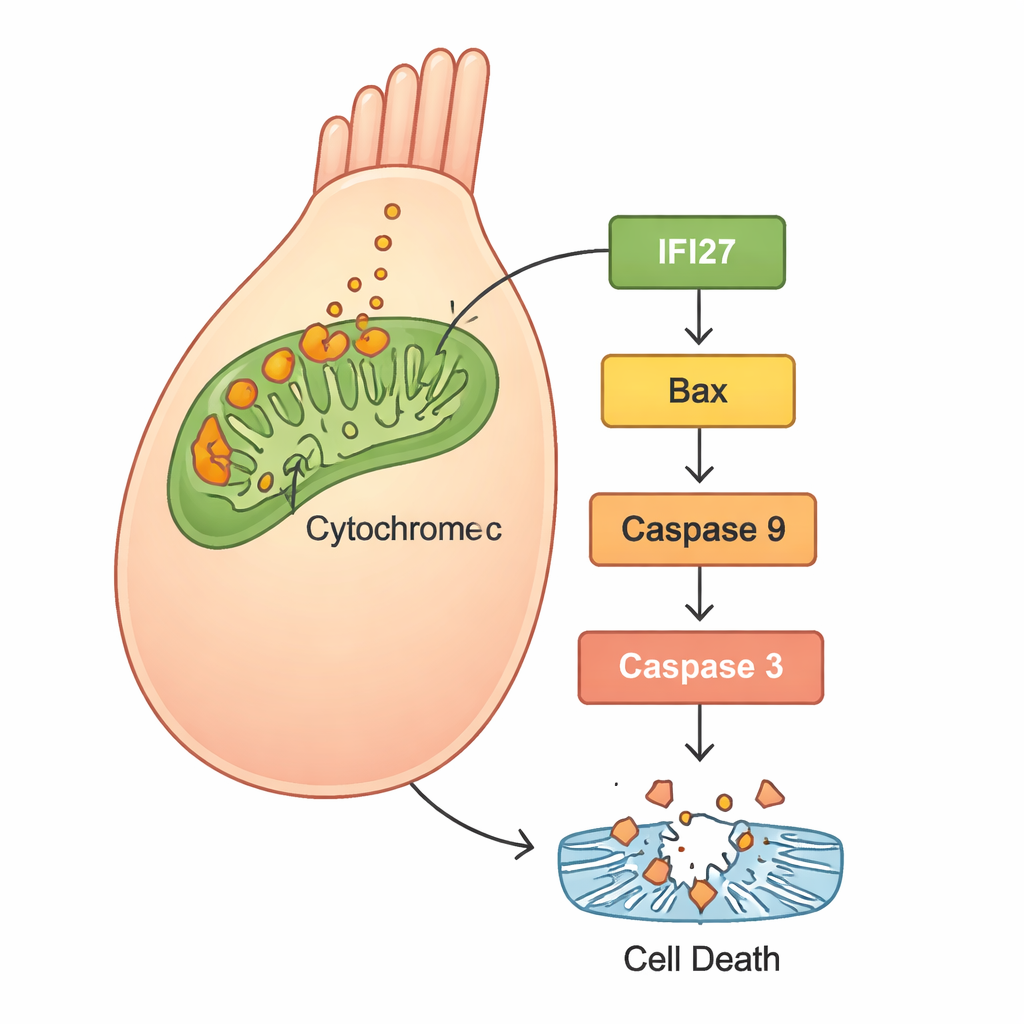
To understand what was happening inside the cells, the scientists used RNA sequencing to compare gene activity between normal zebrafish and those with reduced gjb2. A set of genes linked to the “mitochondrial apoptosis pathway”—a self-destruct route centered in the cell’s energy factories—was strongly activated. In particular, several members of the IFI27 family stood out, along with well-known cell-death players such as Bax, cytochrome c, Apaf1, and caspases. Follow-up experiments in human HEK293 cells confirmed the pattern: cells with the mutant GJB2 produced more reactive oxygen species (ROS, a form of oxidative stress), released more cytochrome c from mitochondria, and switched on apoptosis proteins, leading to increased cell death. When the researchers silenced IFI27 in cells carrying the mutant gene, ROS levels dropped, the death signals were dampened, and fewer cells underwent apoptosis.
What this means for future treatments
Taken together, the findings suggest a clear storyline: the GJB2 c.109G>A mutation disrupts normal inner ear development and function, not only by altering cellular communication, but also by triggering stress inside mitochondria. This stress ramps up IFI27 and related genes, releases cytochrome c, and activates a cascade of proteins that push hair cells toward programmed death. Because hair cells do not readily grow back in humans, their loss leads to permanent hearing deficits. By showing that dialing down IFI27 can soften this destructive cascade in human cells, the study highlights IFI27 as a promising target for drugs or gene-based therapies. While such treatments are still far in the future—and will likely need to be delivered very early in life—this work offers a concrete molecular roadmap for turning a once-mysterious gene mutation into a potentially preventable cause of childhood deafness.
Citation: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Keywords: genetic hearing loss, GJB2 mutation, zebrafish model, mitochondrial apoptosis, IFI27