Clear Sky Science · en
Targeting the Akt–EphA2 axis and cell–cell adhesion enhances anoikis sensitivity in cancer cells
Why free‑floating cancer cells matter
When cancer cells break away from a tumor and travel through the bloodstream, they should normally die because they lose their grip on the surrounding tissue. This built‑in safety program, called detachment‑induced cell death, helps keep healthy tissues from growing where they should not. Metastatic cancer cells, however, often learn to ignore this signal and stay alive while floating, making it easier for them to seed new tumors. This study asks a deceptively simple question: if we restore a key mechanical “sense of touch” in aggressive breast cancer cells, can we force these wandering cells to die, and if not, what backup tricks do they use to survive?
Reawakening a lost sense of touch
Cells constantly test how stiff their surroundings are, tugging on the structures that anchor them. In healthy tissue, losing contact or landing on a very soft surface tends to trigger self‑destruction. The protein Tropomyosin 2.1 (Tpm2.1) is an important part of this internal tension machinery and is often missing or reduced in cancer cells. The researchers used a widely studied metastatic breast cancer cell line and engineered it to produce more Tpm2.1, restoring much of this rigidity sensing. When these modified cells were forced to grow in special non‑stick dishes that keep them from attaching, they did undergo more cell death, grew more slowly, and moved less than unmodified cancer cells. Yet even after several days in suspension, roughly 70% of the Tpm2.1‑expressing cells were still alive, suggesting that reviving their sense of touch alone does not fully solve the problem.
Reading the survival playbook of cancer cells
To understand why some cells died while others lived, the team separated the floating Tpm2.1‑expressing cells into two groups: those showing early signs of cell death and those that appeared healthy. They then measured which genes were turned on or off in each group over four days. The dying cells showed a broad shut‑down of programs involved in cell division, DNA repair, and attachment, along with stress signals that looked like a one‑way road to self‑destruction. By contrast, the surviving cells activated a coordinated survival plan. Early after detachment, they turned on inflammatory and immune‑related pathways and later ramped up major growth and survival circuits, including a well‑known pathway centered on the protein Akt. At the same time, they strongly boosted genes that help cells stick to one another, allowing them to form protective clusters even without gripping the surrounding matrix.
Strength in numbers: how cell clusters resist death
Because the gene data pointed to increased cell–cell adhesion, the researchers tested whether simple crowding could help cells survive in suspension. When Tpm2.1‑expressing cells were grown at high density, where clustering was easy, far fewer cells died than when they were kept sparse. One adhesion molecule, ICAM1, stood out as especially elevated in the surviving cells and is already known to help clusters of circulating tumor cells form and lodge in distant tissues. Blocking ICAM1 with a small‑molecule inhibitor made more of the Tpm2.1‑expressing cells die in suspension, while having little effect on standard attached cultures. These findings support the idea that, once their internal mechanics are partly restored, cancer cells can still escape death by literally holding on to one another instead of to their environment.
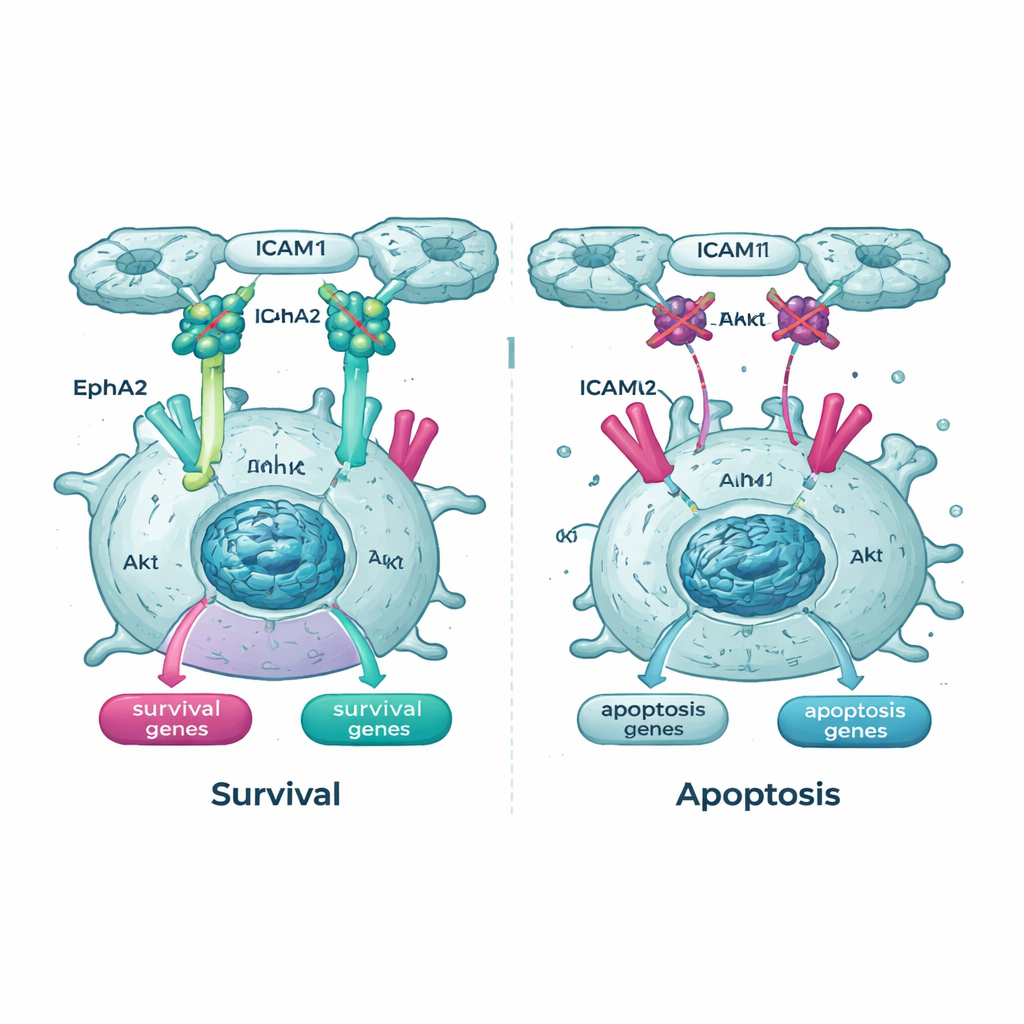
Cutting off key lifelines inside the cell
The genetic surveys also highlighted the Akt pathway and one of its partners, a receptor called EphA2, as important lifelines for suspended cancer cells. In Tpm2.1‑expressing cells, the active, phosphorylated form of Akt was lower in suspension than in control cells, and EphA2 levels and activity were reduced as well. When the team treated cells with drugs that block Akt or EphA2, both normal and Tpm2.1‑expressing cancer cells became much more likely to die in suspension. Notably, the cells with restored rigidity sensing were especially sensitive to these drugs over time, implying that once their mechanical safety checks are partly re‑engaged, they depend more heavily on the remaining survival signals.
Turning a primed state into a lethal weakness
To a non‑specialist, the key message is that fixing one broken safety switch in cancer cells—their ability to feel and respond to losing contact—is necessary but not sufficient to make them self‑destruct. The cells that survive do so by banding together and rerouting their internal wiring through alternative growth and survival pathways. This work shows that restoring rigidity sensing with Tpm2.1 puts metastatic breast cancer cells into a “primed” state where they are closer to death but not yet committed. By then blocking the Akt–EphA2 pathway and disrupting cell–cell adhesion via ICAM1, researchers can push these primed cells over the edge and greatly increase detachment‑induced cell death. In practical terms, the study outlines a strategy for future combination therapies aimed at wiping out free‑floating tumor cells before they can seed new metastases.
Citation: Vivante, A.G., Dwivedi, N., Sheetz, M.P. et al. Targeting the Akt–EphA2 axis and cell–cell adhesion enhances anoikis sensitivity in cancer cells. Sci Rep 16, 6197 (2026). https://doi.org/10.1038/s41598-026-37327-y
Keywords: breast cancer metastasis, cell adhesion, anoikis, Akt signaling, rigidity sensing