Clear Sky Science · en
Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data
Why this matters for gene research
Modern gene-expression tests can measure tens of thousands of genes in a single patient sample. That flood of data promises earlier cancer diagnosis and better treatment choices, but it also creates a problem: most of those genes are noisy, redundant, or tied mainly to common cases, not the rare and dangerous ones. This paper presents a new way to sift through massive gene-expression datasets so that computers can reliably spot patients in a small, hard‑to‑detect minority group using only a tiny, carefully chosen set of genes.
The challenge of too many, too similar genes
Microarray experiments often track thousands of gene activity levels for only a few hundred patients. Usually, one class (such as a common cancer subtype) greatly outnumbers the other, creating highly imbalanced data. In this setting, many genes behave in very similar ways, and the patterns for majority and minority patients can overlap. Standard learning methods tend to latch onto the majority class and to be confused by redundant genes, which leads to overfitting and poor detection of rare subtypes. Traditional dimensionality‑reduction methods either throw away interpretability by building new mixed features, or select genes without looking closely at how well they help a classifier recognize the minority cases.
A new roadmap for smarter gene selection
The authors introduce AFCG‑SFE, an adaptive feature‑selection model designed specifically for high‑dimensional, imbalanced gene-expression data. The method starts from a simple “single‑agent” search that turns genes on or off and tests how well they support classification, but enriches it with several data‑driven steps. First, it groups genes based on how similarly they behave, then lets genes belong to more than one group to reflect the biological reality that a gene may be involved in multiple pathways. Inside each group, it ranks genes by how informative they are about the disease label and keeps only a few key representatives, sharply cutting redundancy before the main search even begins. 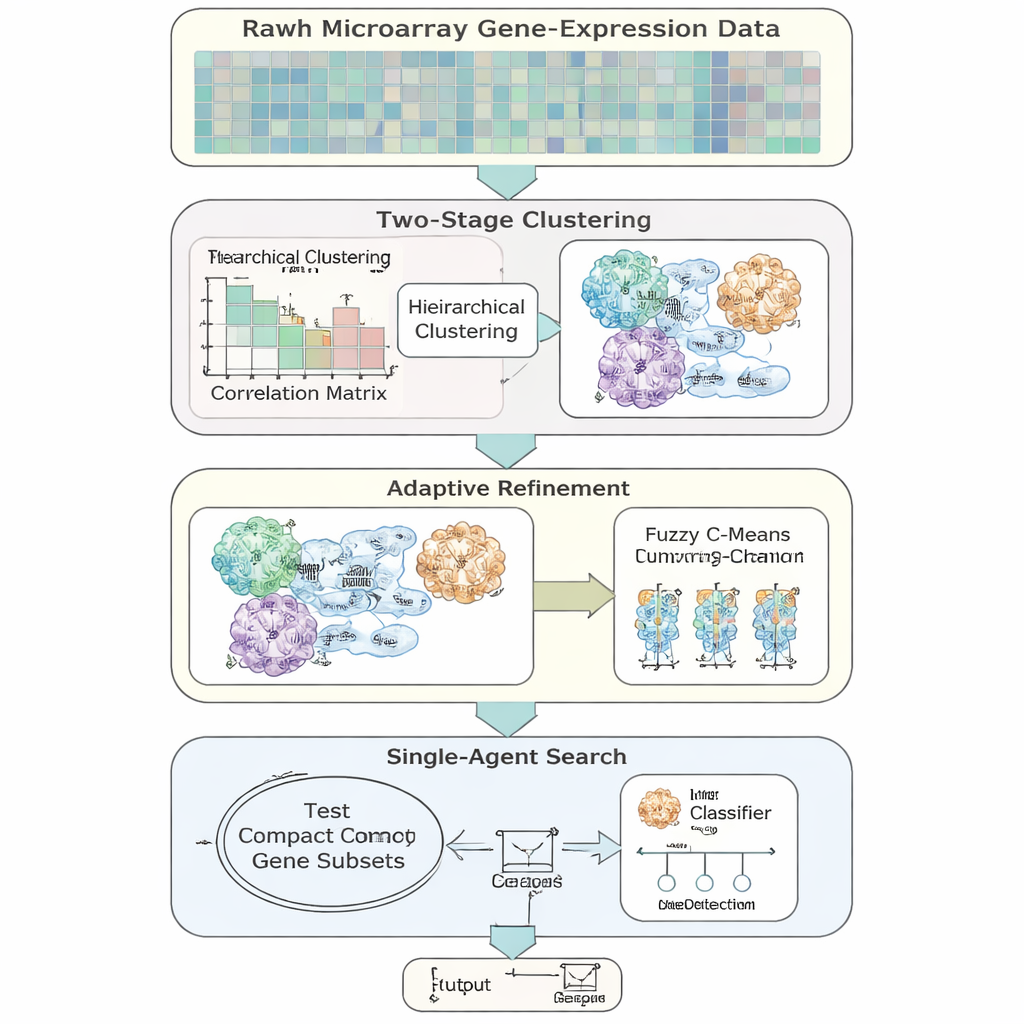
Making the computer care about rare patients
Instead of focusing on plain accuracy, AFCG‑SFE uses a fitness score that emphasizes metrics suited to skewed data, including the balance between correctly identifying minority and majority cases and performance across all decision thresholds. The fitness function also includes penalties for selecting too many genes or many genes from the same cluster, and a reward for genes that carry strong dependence on the disease label. Importantly, the strength of these penalties and rewards is set automatically from properties of the dataset, such as how many genes there are per patient and how much the classes overlap, rather than by manual tuning. This makes the method more robust and easier to transfer between studies.
Adapting to problem difficulty
A key idea is that the algorithm should not always aim for the smallest possible gene set. When the two classes are very hard to separate or heavily overlapped, the method automatically raises a lower bound on how many genes must be kept, ensuring that rare but important signals are not thrown away. As the search progresses, AFCG‑SFE gradually tightens a per‑cluster cap on how many genes can survive from each group, while still respecting this minimum. The result is a compact, diverse panel of genes that captures the structure of the data without being dominated by a single, redundant pattern. 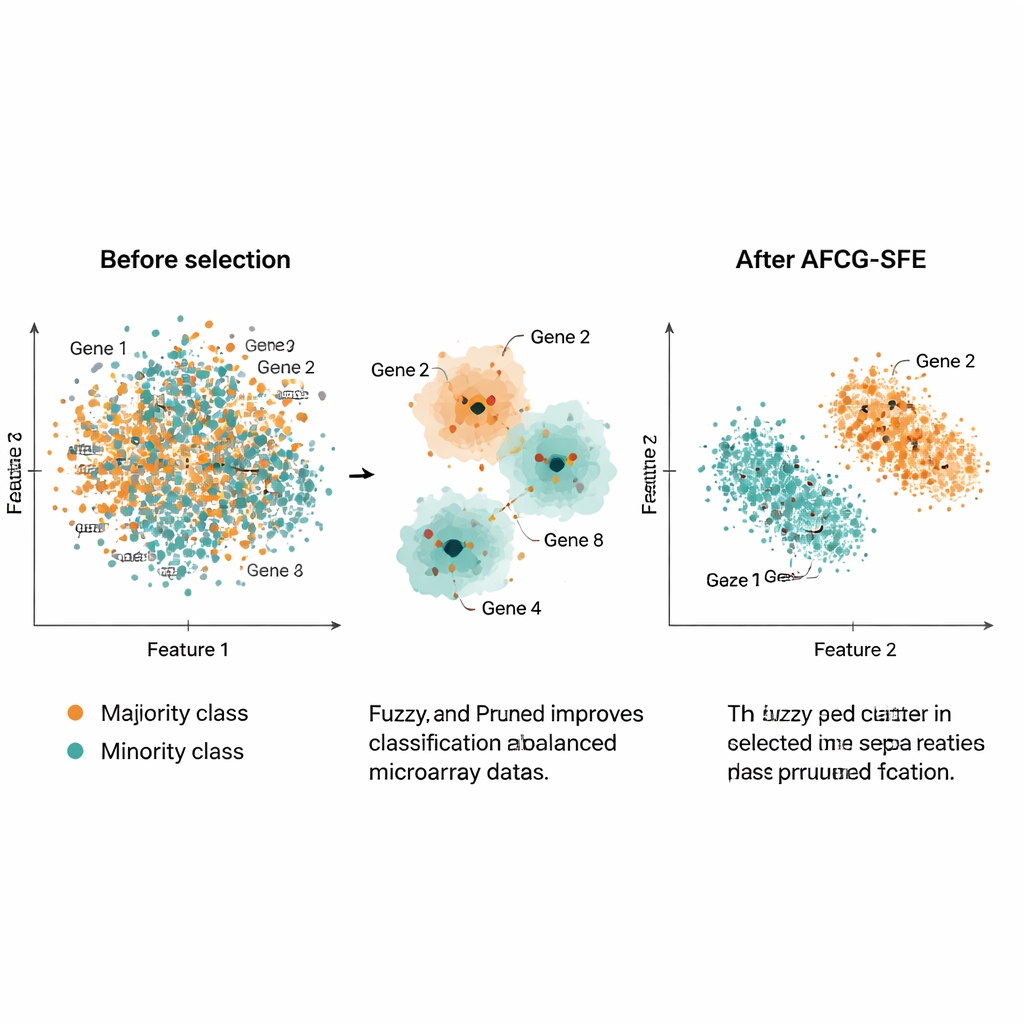
What the experiments show
The authors tested AFCG‑SFE on 20 public cancer microarray datasets, each with thousands of genes but only about 100–200 samples and strong class imbalance. They compared their method with several evolutionary search baselines, simple filters, and embedded approaches that build feature selection into the classifier. Across a battery of measures—including F‑measure, balanced accuracy, area under the ROC curve, and a measure of overfitting—AFCG‑SFE was best or tied for best on all datasets. It typically selected fewer than 25 genes (often as few as 6–8), removing more than 99% of the original features while still improving or maintaining classification performance. It also reduced a complexity index that captures how much the classes overlap in feature space, indicating clearer separation after selection.
Bottom line for non‑experts
In practical terms, this work offers a way to shrink huge, noisy gene‑expression profiles down to very small sets of informative genes that still let computers accurately recognize rare patient subgroups. By intelligently grouping similar genes, rewarding those that truly track the disease, and explicitly guarding against bias toward the majority class, AFCG‑SFE delivers both better prediction and much simpler gene panels. That combination can help researchers home in on potential biomarkers, design more interpretable diagnostic tests, and ultimately improve how precision medicine tools work with real, imperfect biological data.
Citation: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Keywords: gene expression, feature selection, imbalanced data, microarray, cancer subtypes