Clear Sky Science · en
rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences
Why tiny cold viruses still matter
Most of us think of the common cold as a nuisance rather than a serious threat. Yet the viruses that cause many colds—human rhinoviruses—are also linked to severe lung infections, asthma attacks, and flare‑ups of chronic lung disease. To track how these viruses evolve and spread, scientists need to sort them into precise genetic “types,” much like assigning barcodes to products. This article introduces rhinotypeR, a free, open‑source software package that makes this genetic labeling more accurate, consistent, and easy to repeat, helping public health teams keep a clearer eye on an often‑overlooked family of respiratory viruses.
The hidden variety in common colds
Human rhinoviruses are extraordinarily common, showing up in as many as 60% of samples from people with sudden respiratory illness. Far from being a single virus, they are split into three main groups, called A, B, and C, and at least 169 recognized genetic types. Different types behave differently: some are more often tied to severe infections in children and to asthma flare‑ups, while others are seen less frequently in serious disease. Because these types evolve independently and carry distinct surface features, scientists need reliable ways to tell them apart if they are to follow how outbreaks move through schools, households, and communities.
From scattered tools to one clear path
Until now, assigning a rhinovirus type from its genetic code has been a patchwork job. Researchers typically focused on a short stretch of the virus’s genome called the VP4/2 region, lined it up with known reference strains, measured how different the sequences were, and then applied cut‑off values to decide which type each sample belonged to. But these steps were carried out with a mix of software programs, manual edits, and personal judgment. That made it hard for different studies to be compared or repeated, even when they used similar data. rhinotypeR was created specifically to turn this multi‑step, error‑prone process into a single, scripted workflow that anyone can run and share.
What the new software actually does
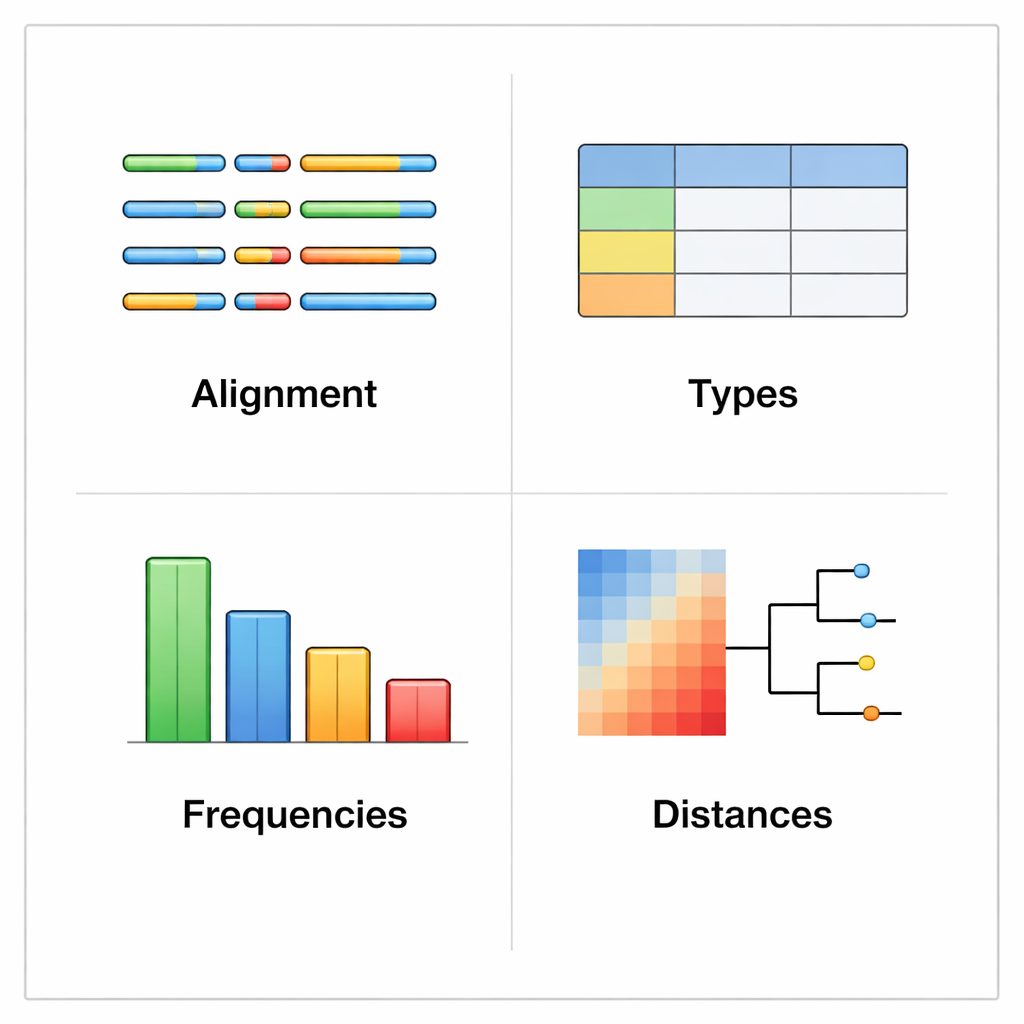
rhinotypeR runs inside the widely used R and Bioconductor environment for data analysis. It takes in a collection of rhinovirus VP4/2 sequences and walks them through three main stages: preparing and aligning the sequences, calculating how distant each one is from a curated set of reference types, and then assigning each sample to the closest known type or flagging it as “unassigned” if it is too different. The same tool can produce visual outputs, including color‑coded maps of genetic differences, simple family trees, and charts showing how common each type is in a dataset. Users can align their data with external programs if they prefer, or let rhinotypeR handle the entire process within R for maximum reproducibility.
Putting the tool to the test
To check that rhinotypeR gives trustworthy results, the authors compared its distance measurements with those from two established programs, ape and MEGA X, using the same input files and models. The results matched almost perfectly; any tiny discrepancies were down to normal rounding in computer arithmetic, not real differences in method. The team then ran rhinotypeR on a large collection of more than 2,300 rhinovirus sequences from multiple earlier studies, covering over 90% of known types. In roughly four out of five cases, the new tool agreed exactly with previous type labels. Most disagreements occurred right around the pre‑agreed cut‑off points used to separate one type from another, which is precisely where borderline calls are most expected. Importantly, samples that could not be confidently assigned to a known type did not show signs of simply being poor‑quality or low‑virus specimens, suggesting they may reflect genuine viral diversity.
Why this matters for public health
For non‑specialists, the key message is that rhinotypeR does not reinvent how scientists classify cold viruses; instead, it makes that process clearer, more transparent, and easier to repeat. By bundling alignment, distance calculations, and type assignment into one open‑source package—along with clear visual summaries—it helps researchers and surveillance programs process thousands of samples in a consistent way. That consistency improves our ability to compare studies from different places and times, spot unusual or emerging virus lineages early, and link genetic patterns to real‑world disease trends. In the long run, tools like rhinotypeR strengthen routine monitoring of seemingly ordinary colds that, in many people, can trigger serious illness.
Citation: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Keywords: rhinovirus genotyping, molecular surveillance, VP4/2 sequencing, bioinformatics tools, respiratory viruses