Clear Sky Science · en
Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods
Why this matters for cancer care
When doctors and scientists study cancer, they often turn to the cell’s “message molecules” — RNA — to see which genes are active or silent. These patterns can reveal how a tumor behaves and which treatments might work best. But most hospital samples are stored in wax blocks after being treated with formalin, which damages fragile RNA. This study asks a practical question with big consequences for cancer research: now that a widely used RNA test has disappeared from the market, can a newer method step in and deliver equally useful results from these routine preserved samples?
Two tools for reading gene activity
For years, many labs relied on a method called HTG EdgeSeq Human Transcriptome Panel (HTP) to read gene activity directly from tiny scrapings of formalin-fixed, paraffin-embedded (FFPE) tissue. This approach could survey almost all human genes without first extracting RNA, saving time and preserving precious material. However, the company behind HTG EdgeSeq went bankrupt, leaving researchers searching for an alternative. A newer technology, TempO-Seq (TOS), from another manufacturer, promises similar abilities: it also targets many genes at once, works on damaged RNA from FFPE samples, and is designed to be sensitive, reproducible, and relatively affordable.
Putting the methods to the test
The research team compared these two technologies head-to-head in a very practical setting. They analyzed 21 stored endometrial cancer samples, along with three standard RNA reference materials, first with HTG HTP and then with TempO-Seq. Both methods used targeted panels that together covered more than 18,000 of the same genes. The scientists applied strict quality checks, ensuring that each sample produced enough sequencing reads and that the measurements were stable. They also used statistical tools to remove “batch effects” — artificial differences that can arise simply because tests were run on different days, machines, or platforms.
What matches and what does not
When the team looked at the expression of individual genes one by one, the two methods did not always agree. Differences in how each technology designs its probes, prepares samples, and counts reads can make single-gene comparisons noisy. However, this picture changed when they examined broader patterns that combine information from many genes at once. Multi-gene signatures — such as those used to group tumors into molecular subtypes, estimate how many immune cells are present in a sample, or guess how pure the tumor tissue is — showed much stronger agreement between TempO-Seq and HTG. In most cases, the scores or classifications were similar, even after the researchers simulated using fewer sequencing reads to mimic different machine capacities.
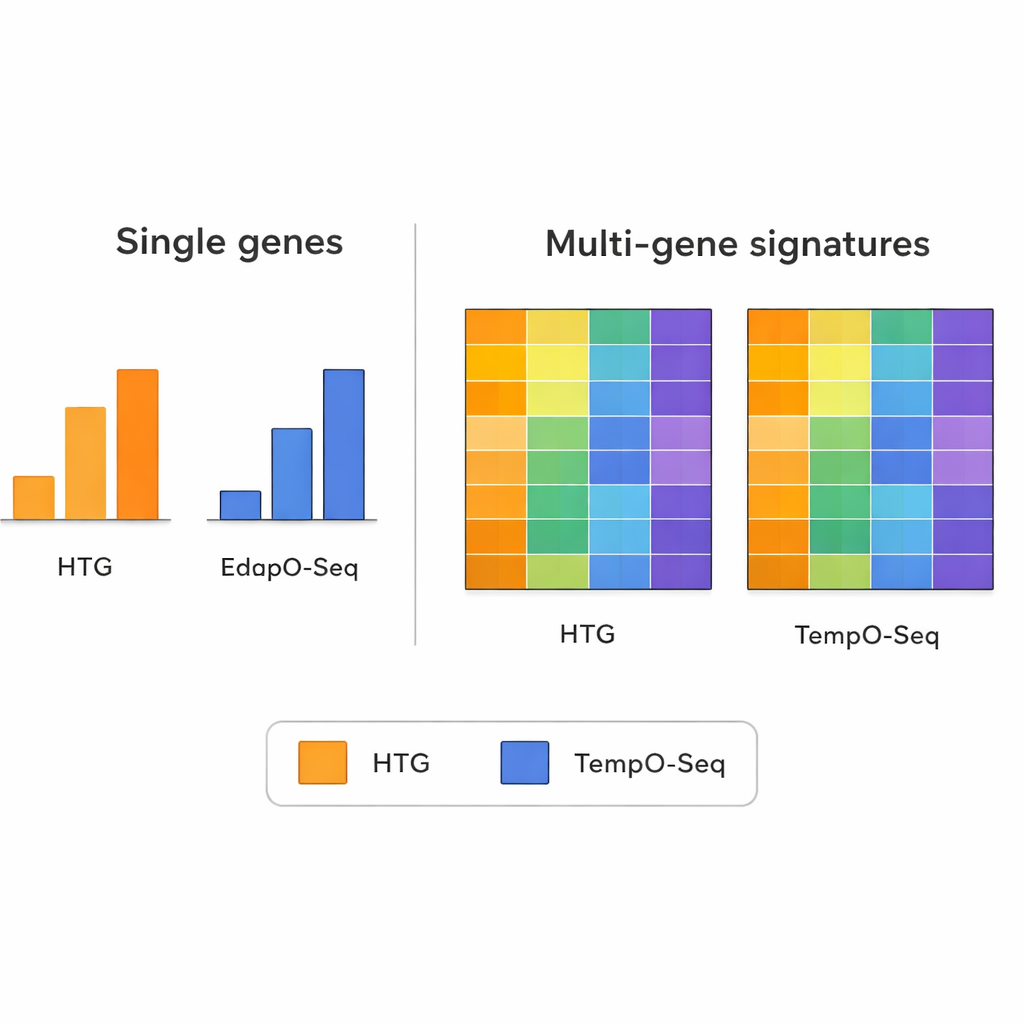
Multi-gene patterns as reliable signals
The study highlights an important principle in modern genomics: while any single gene’s measurement can be thrown off by technical quirks, combining signals from dozens or hundreds of genes tends to average out that noise. The authors used several widely known multi-gene tools as technical stress tests. These included a breast cancer panel that assigns tumors to intrinsic subtypes, an algorithm that scores how much immune and connective tissue is mixed into a tumor sample, and a method that estimates the proportions of many types of immune cells. Across these complex readouts, TempO-Seq usually tracked closely with HTG, suggesting that it captures the same biological stories even if some of the fine-grained details differ.
What this means going forward
For researchers who depend on FFPE archives to study cancer, the loss of a trusted platform could have been a major setback. This benchmarking study offers reassurance: TempO-Seq appears to be a solid replacement for HTG HTP when the goal is to use multi-gene biomarkers and broad expression patterns, which are the backbone of many modern diagnostic and prognostic tools. The authors caution that directly comparing single-gene results across platforms is unwise, because each method targets genes in slightly different ways. Instead, they recommend focusing on complex, multi-gene signatures for cross-platform work. In plain terms, the new method seems capable of carrying on the job of its predecessor for most real-world oncology research needs, especially when scientists care about the overall pattern of many genes rather than the exact value of just one.
Citation: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Keywords: transcriptomic profiling, endometrial cancer, FFPE tissue, targeted RNA sequencing, gene expression biomarkers