Clear Sky Science · en
A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts
Everyday Chemicals with Hidden Complexity
From fabric softeners and hair conditioners to disinfectant wipes and mouthwash, a family of chemicals called quaternary ammonium salts—often shortened to “Quats”—quietly powers many products we rely on. They help kill germs, soften clothes, and speed up industrial reactions. Yet predicting exactly how these salts behave in water has been surprisingly difficult, limiting how efficiently we can design safer, greener formulations. This study shows how modern machine learning can learn from past measurements to predict that behavior more flexibly and, in many cases, more accurately than traditional models.
Why These Salts Matter
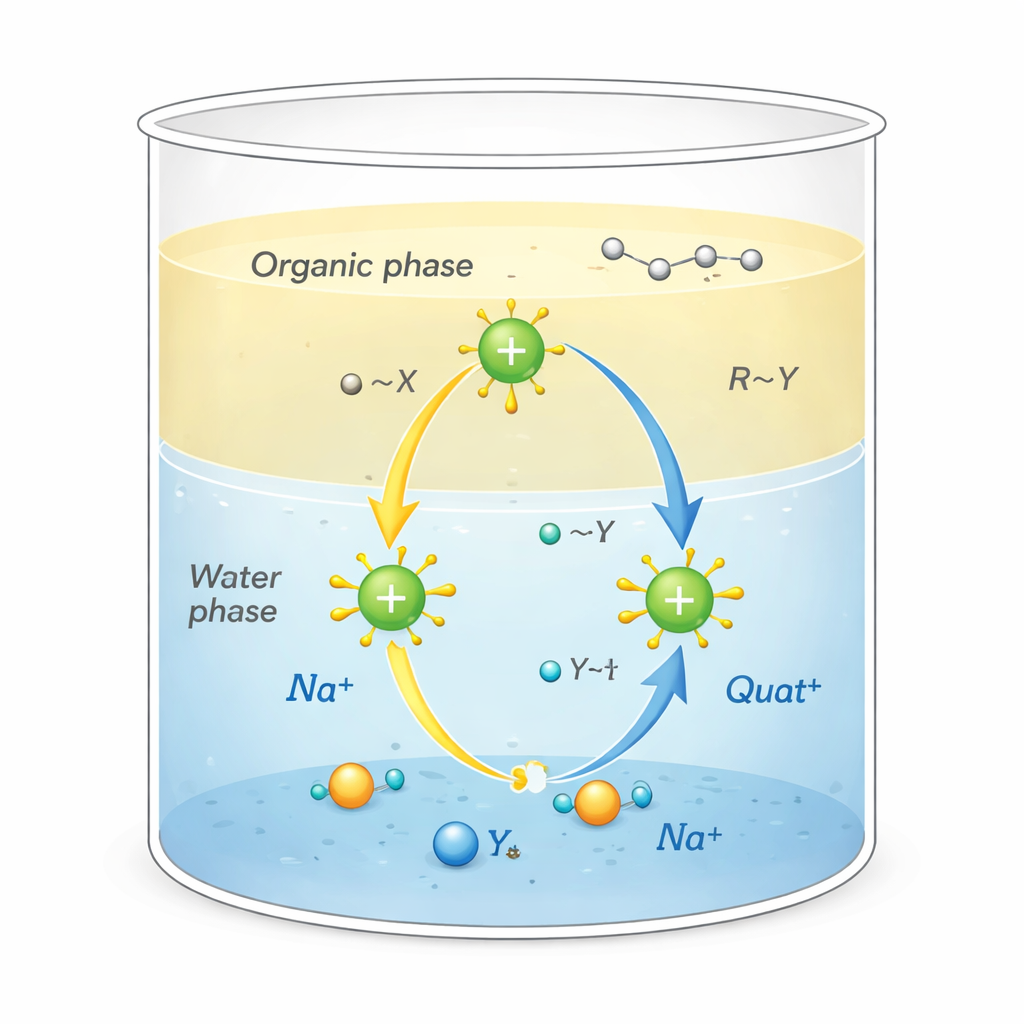
Quats are positively charged molecules surrounded by carbon‑rich “tails.” This unusual shape allows them to do several jobs at once: stick to oily dirt, cling to surfaces such as fabric or hair, and disrupt the membranes of microbes, making them powerful disinfectants and surfactants. They are also used as phase‑transfer catalysts, acting like shuttles that carry reactive ions from water into oil‑like solvents where they normally would not go. That shuttling action, which takes place at the boundary between water and oil, can dramatically speed up chemical reactions used in manufacturing pharmaceuticals, polymers, and fine chemicals.
Why It Is Hard to Predict Their Behavior
To design new Quats or tune existing ones, chemists need to know how they behave in solution—how strongly they interact with water and with other dissolved ions. Two key measures are the osmotic coefficient, which reflects how salts affect the tendency of water to draw across membranes, and the activity coefficient, which captures how “effective” a dissolved species is compared with an ideal, perfectly mixed solution. Traditionally, these values are obtained either by painstaking experiments or by using complex physical models such as Electrolyte‑NRTL and Extended UNIQUAC, which require many fitted parameters and are not easy to generalize to new molecules.
Teaching a Computer to Read Molecules
The researchers took a different route: they asked whether a computer could learn the link between Quat structure and osmotic behavior directly from existing data. They gathered 1,654 measurements of osmotic coefficients for 52 different Quats from the scientific literature. Each molecule was described using SMILES notation—a string representation that encodes features such as the number of carbon and oxygen atoms, the presence of benzene rings, branching, and the type of positively charged nitrogen group, along with the accompanying negative ion (like chloride, bromide, or nitrate). These structural descriptors, plus the salt concentration, served as inputs to several supervised machine‑learning algorithms implemented in Python.
Finding the Most Reliable Predictor
Seven different algorithms, including linear regression, decision trees, random forests, support vector machines, gradient boosting, k‑nearest neighbors, and Gaussian processes, were trained on 70% of the data and tested on the remaining 30%. The team also used a stricter validation scheme in which all data for one salt were held out to see how well the models extrapolated to a truly unseen compound. Linear regression performed poorly, missing important nonlinear trends. Tree‑based methods fit the training data extremely well but produced slightly jagged predictions and lost accuracy on new salts. The Gaussian process model struck the best balance: it delivered smooth, physically reasonable curves for osmotic coefficients and achieved a mean absolute percentage error of about 5% overall, outperforming alternative machine‑learning approaches under the toughest tests.
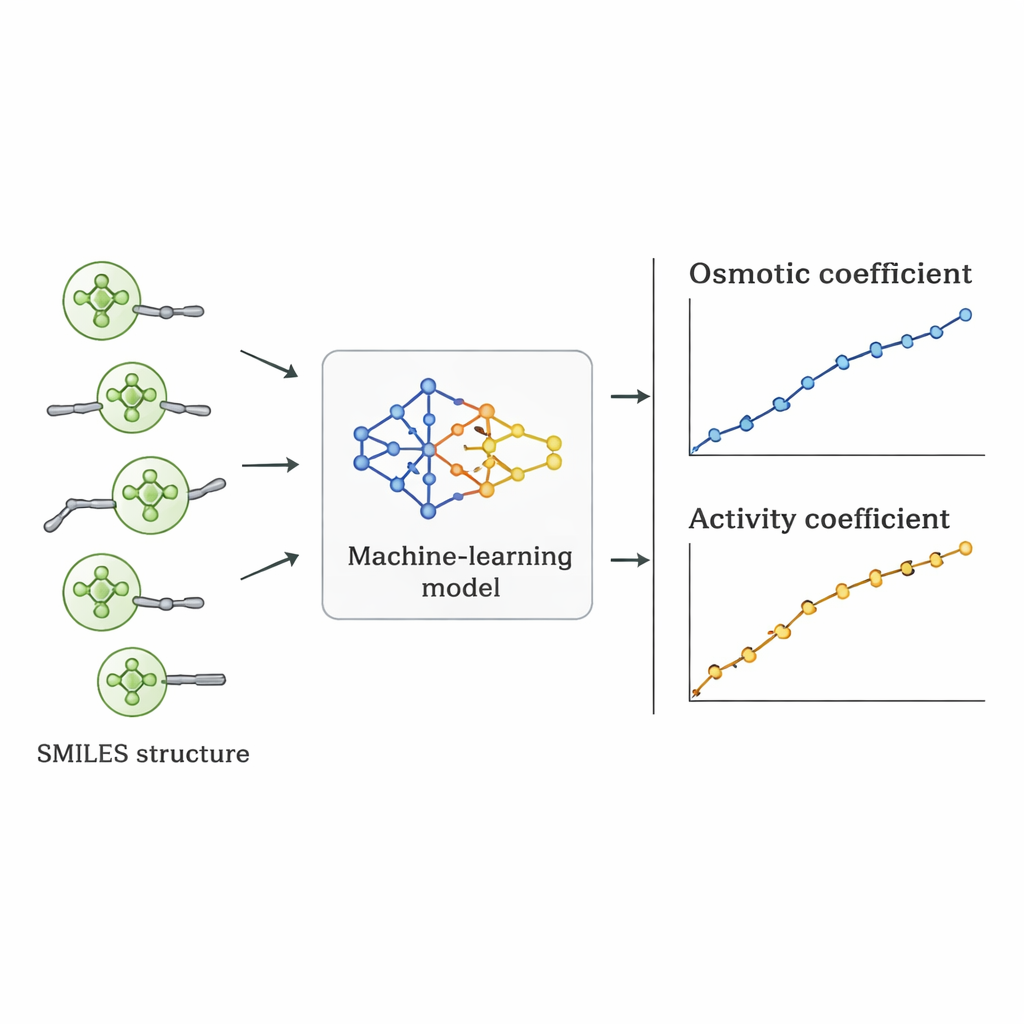
From Osmotic Behavior to Useful Design Numbers
Once the best model was chosen, its predicted osmotic coefficients were converted into activity coefficients using standard thermodynamic relationships. When these activity coefficients were compared with values derived from experiments and from established physical models, the machine‑learning approach often matched or beat them for individual Quats. Although its average error across all substances was slightly higher than some specialized models, it had a crucial advantage: because it is driven by structural descriptors rather than salt‑specific fitting, it can be applied to new Quats that have never been measured in the lab, as long as their structures resemble those in the training set.
What This Means for Products and Processes
For a non‑specialist, the message is that computers can now “read” compact text descriptions of molecules and, from patterns learned in past data, predict how those molecules will behave in water with impressive accuracy. This opens the door to faster, cheaper screening of new Quats for disinfectants, cleaners, personal‑care products, and industrial catalysts, without exhaustive experimentation for every candidate. The current model is just a first step, and the authors note that richer molecular fingerprints and newer algorithms could further improve performance. Still, it demonstrates how data‑driven tools can complement traditional chemistry, helping engineers design more effective and potentially safer formulations by exploring chemical possibilities that would be impractical to test one by one in the lab.
Citation: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Keywords: quaternary ammonium salts, phase-transfer catalysis, osmotic coefficients, activity coefficients, machine learning in chemistry