Clear Sky Science · en
NEK7 phosphorylation of cortactin modulates the migratory capacity of cells expressing EML4-ALK V3
Why cell movement matters in lung cancer
Cancer becomes life-threatening when cells learn to travel. In non-small cell lung cancer, some tumors spread through the body faster than others, and one culprit is a faulty fusion protein called EML4-ALK. A particular version, known as variant 3 (V3), is linked to especially aggressive disease and poor responses to targeted drugs. This study asks a basic but vital question: what lets these V3-driven cancer cells change shape and move so efficiently, and can we pinpoint the molecular parts that make this possible?
A hyper‑migratory lung cancer variant
Doctors have long known that only a minority of lung cancers carry the EML4-ALK fusion, but patients whose tumors express the V3 form tend to do worse than those with other variants. Under the microscope, V3-expressing cells look different: instead of being compact and cobblestone-shaped, they stretch out into long, thin forms with extended protrusions, resembling cells on the move. Earlier work showed that this behavior depends on two enzymes, NEK9 and NEK7, which act as molecular switches in cells. However, the crucial downstream targets of these switches—those that directly reshape the cell’s internal skeleton—were not well understood.
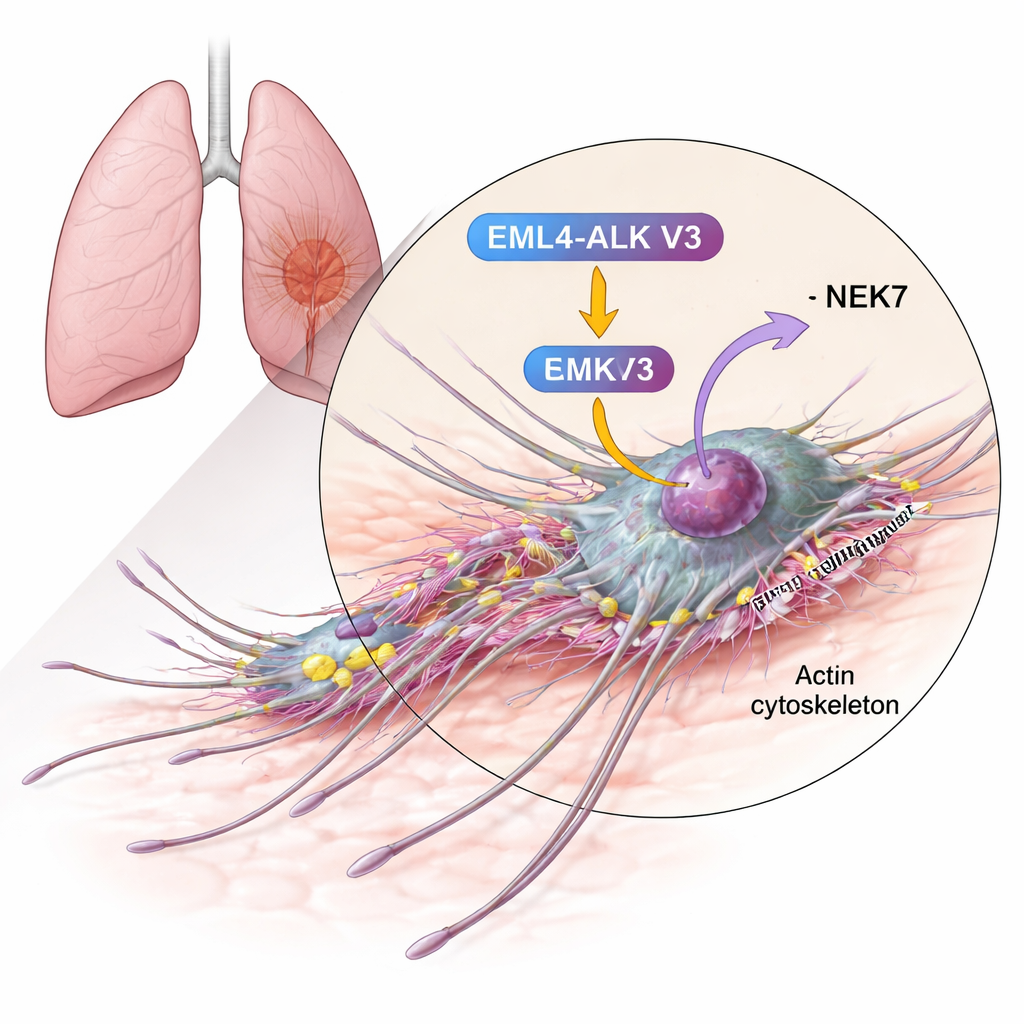
Linking a movement protein to an aggressive fusion
The authors homed in on cortactin, a protein already known to be abundant in many invasive cancers and to help build the networks of actin filaments that push the cell membrane forward. Using biochemical tests, they showed that cortactin can be chemically modified (phosphorylated) by NEK6 and, even more strongly, by NEK7. The enzymes add phosphate groups to specific serine residues within cortactin’s actin-binding region—the very stretch that grips actin fibers and stabilizes branched networks. When NEK7 was present, cortactin was decorated with more phosphate groups and at more sites than with NEK6, suggesting NEK7 is the primary regulator in this context.
Turning off cortactin stalls cancer cell migration
To see what cortactin actually does in living cells, the researchers depleted it using RNA interference in cells engineered to activate NEK9 or NEK7, or to express EML4-ALK V3 itself. In all three settings, the striking elongated, mesenchymal-like shapes collapsed: cells became flatter and rounder, lost their long protrusions, and instead formed thick, straight actin “stress fibers” crisscrossing the cell. Multiple migration assays—from closing artificial “wounds” in a cell sheet to tracking single cells and measuring movement toward a chemical signal—showed that without cortactin, these highly motile cells slowed down dramatically. Similar effects were observed in established lung cancer cell lines naturally carrying EML4-ALK V3, underscoring the clinical relevance of the pathway.
Fine filaments and sharp tips at the leading edge
High-resolution imaging in bronchial epithelial cells revealed an even more detailed picture. EML4-ALK V3-expressing cells produced many fine, sometimes branched, filopodia-like extensions studding their protrusions. At the tips and branching points of these structures, cortactin, EML4-ALK V3, NEK7, and a phosphorylated form of cortactin all gathered together. This tight co-localization suggests a focused “construction site” where NEK7 modifies cortactin to build and maintain the delicate, branched actin networks that help steer the cell. When cortactin was removed, these intricate extensions disappeared and invasive growth from 3D tumor-like spheroids into a surrounding gel was strongly reduced.
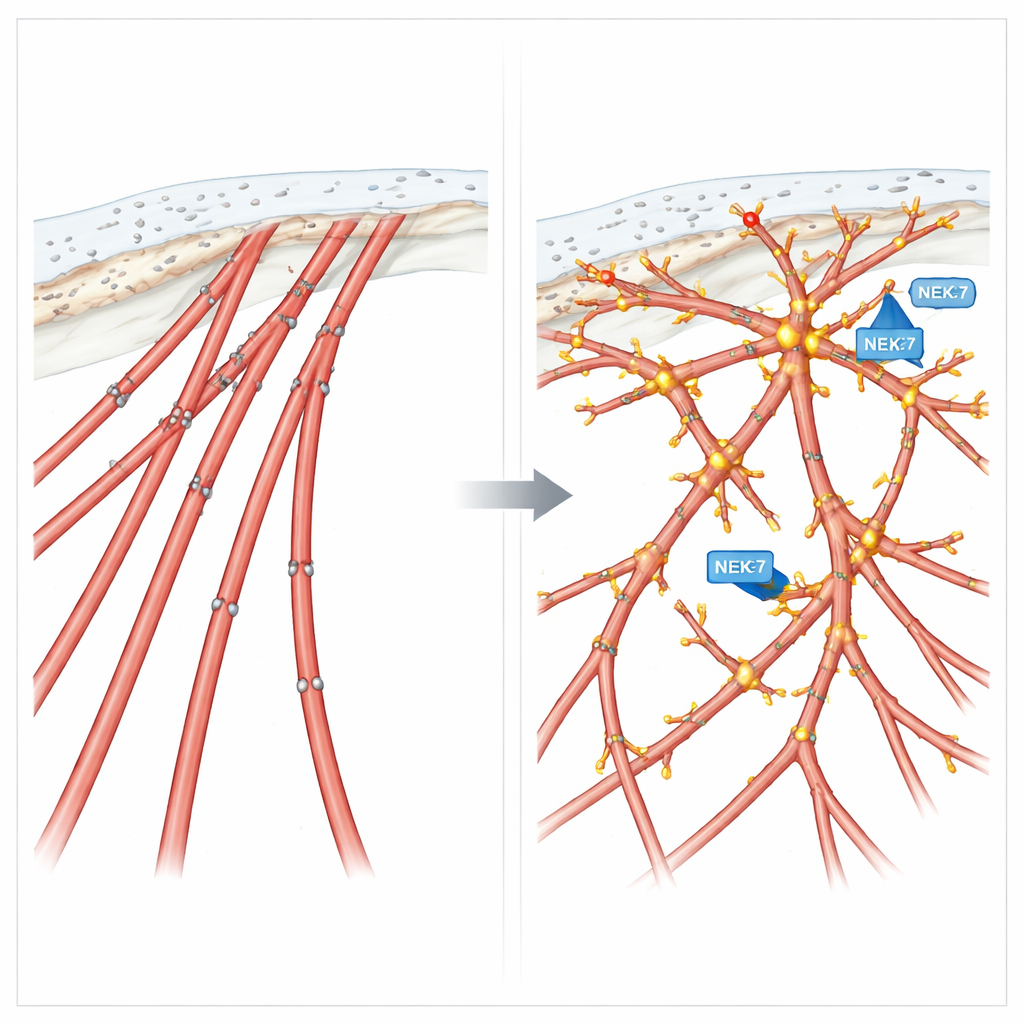
Phosphorylation as a migration dial
To test how these chemical tags on cortactin influence behavior, the team created two designer versions of the protein: a phospho-mimetic form that imitates constant phosphorylation at four key sites, and a phospho-null form that cannot be phosphorylated there. Cells expressing the mimetic version developed abundant filopodia-like extensions and showed enhanced directed migration, similar to cells with active NEK7 or EML4-ALK V3. In contrast, cells expressing the non-phosphorylatable version formed rigid stress fibers, lost those fine extensions, and moved in a fast but aimless way—good at wandering, poor at following a signal. In three-dimensional cultures, this phospho-null cortactin drove invasive outgrowths that were disorganized rather than precisely guided.
What this means for understanding—and targeting—spread
Put simply, the study shows that the aggressive EML4-ALK V3 lung cancer variant hijacks a normal cell-shaping system. By activating NEK7, it causes cortactin to be phosphorylated at specific sites within its actin-binding region. This modification tunes cortactin so it can build finely branched actin structures and filopodia-like extensions that support fast, directed cell migration and invasion. Disrupting cortactin or its phosphorylation flips the system: cells either barely move or move chaotically without direction. These insights reveal a concrete molecular chain—from a cancer-driving fusion, through NEK7, to cortactin and the actin cytoskeleton—that helps explain why some lung cancers metastasize so effectively, and point toward new ways to slow or misdirect their movement.
Citation: Richardson, E.L., Knebel, A., Straatman, K.R. et al. NEK7 phosphorylation of cortactin modulates the migratory capacity of cells expressing EML4-ALK V3. Sci Rep 16, 6407 (2026). https://doi.org/10.1038/s41598-026-36484-4
Keywords: non-small cell lung cancer, EML4-ALK V3, cell migration, cortactin, actin cytoskeleton