Clear Sky Science · en
ATF4 regulates mitochondrial dysfunction and mitophagy, contributing to corneal endothelial apoptosis
Why the window of the eye can turn cloudy
Our corneas—the clear front windows of the eyes—stay transparent thanks to a thin, hard‑working layer of cells on their inner surface. In Fuchs’ endothelial corneal dystrophy (FECD), millions of people slowly lose these cells, leading to swelling, cloudy vision, and often corneal transplants. This study asks a basic but crucial question: what makes these cells decide to die, and could turning off one molecular “switch” help save them?
A fragile cell layer that keeps vision clear
The corneal endothelium is a single sheet of hexagonal cells that constantly pumps fluid out of the cornea to keep it clear. In FECD, these cells become stressed and gradually disappear, while bumps of abnormal material, called guttae, build up on the underlying membrane. Because there are no approved drugs for FECD and corneal transplants are the main treatment, researchers are trying to understand exactly how stress inside these cells pushes them toward death. Earlier work pointed separately to strain in two key cellular compartments—the endoplasmic reticulum (the cell’s protein‑folding factory) and mitochondria (the cell’s power plants)—but how these two stress responses talk to each other remained unclear.
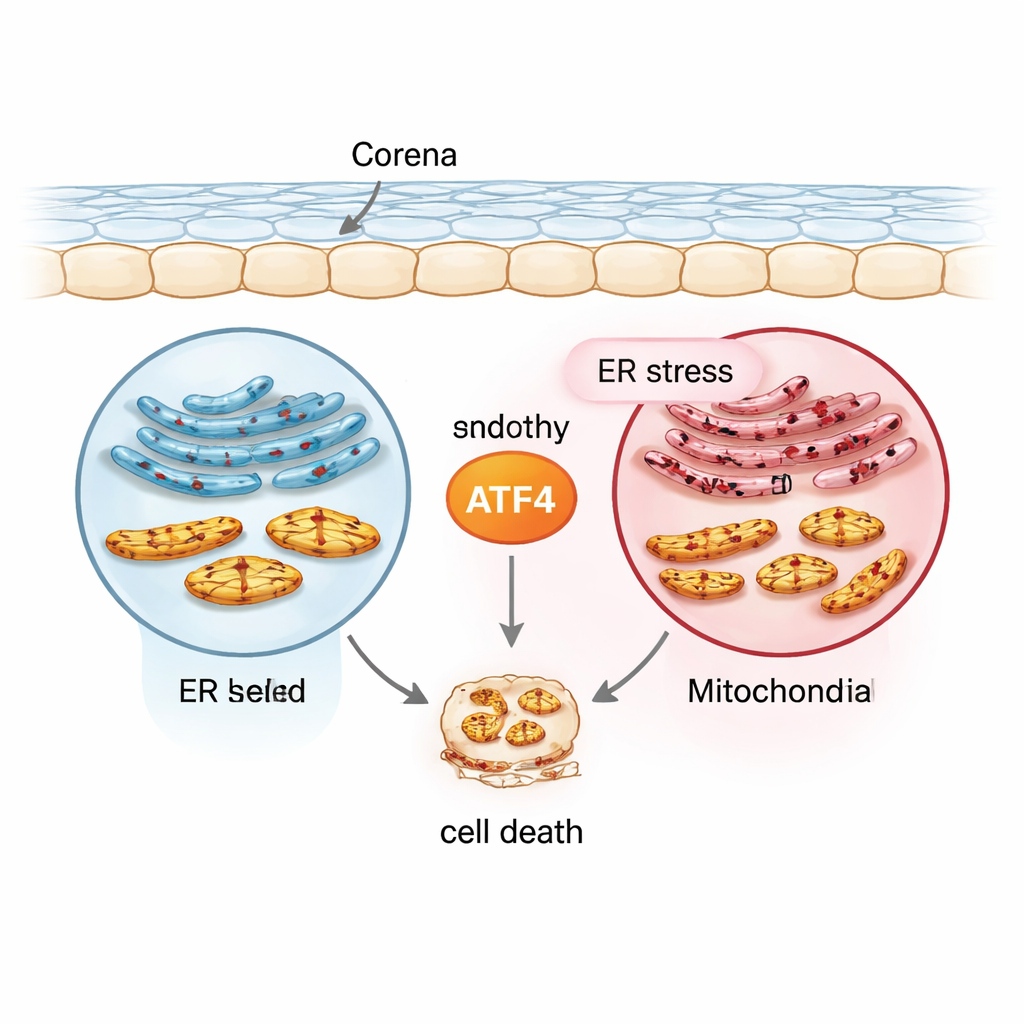
The stress messenger at the center: ATF4
The team focused on a protein called ATF4, a transcription factor that switches many stress‑response genes on or off. Using a normal human corneal endothelial cell line (21T), a FECD‑like cell line carrying the disease‑linked TCF4 repeat expansion (F35T), primary human corneal endothelial cells, and mouse models exposed to ultraviolet A (UVA) light, they created a range of conditions that mimic chronic stress. They triggered endoplasmic reticulum stress with a drug called tunicamycin and then measured ATF4 and other markers. Compared with normal cells, FECD‑like cells started out with higher levels of ATF4 and related stress proteins, and ATF4 rose even further under chronic stress in both cultured cells and human corneal tissues. This pattern placed ATF4 at the crossroads between early protective responses and later, self‑destructive signaling.
From power failure to programmed death
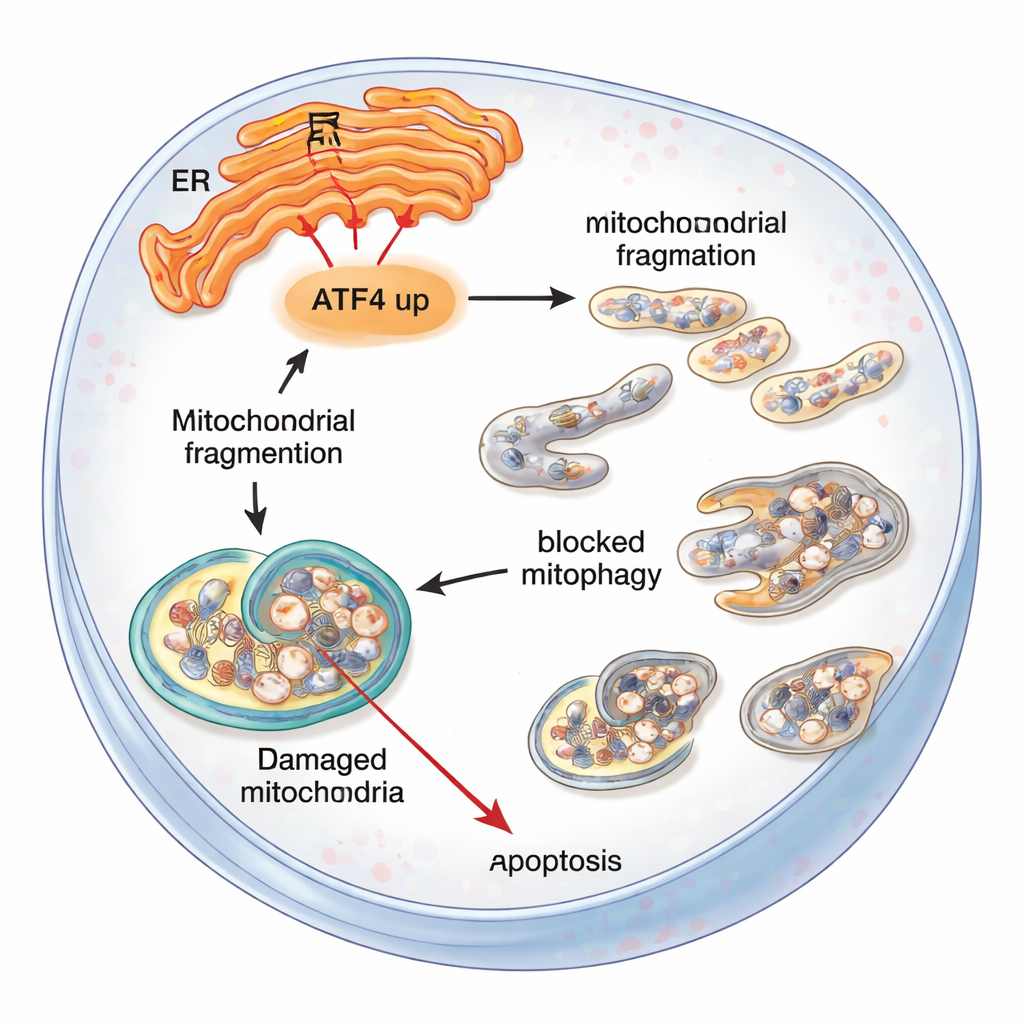
Next, the researchers looked at how this stress affected mitochondria. In FECD‑like cells, mitochondria produced less ATP, lost their electrical membrane potential, and broke from long, network‑like shapes into many small fragments. These changes worsened when endoplasmic reticulum stress was prolonged. At the same time, classic cell‑death proteins—such as activated caspases and the DNA‑repair protein PARP in its cleaved, pro‑death form—became more abundant, while protective proteins like Bcl‑2 dropped. Together, these changes indicate that stressed corneal endothelial cells in FECD are pushed toward mitochondria‑driven apoptosis, a tidy but irreversible form of programmed cell suicide.
Cleanup system stalls under chronic stress
Ordinarily, heavily damaged mitochondria are removed by a recycling process called mitophagy, in which they are tagged and wrapped into small sacs for disposal. The team found that early mitophagy “starter” molecules (Parkin and LC3) were switched on in both normal and FECD‑like cells, especially after stress. But key supporting proteins were reduced, and electron microscopy showed a buildup of partially digested mitochondria trapped in vesicles. This suggested that while the cleanup process started, it failed to finish, leaving cells cluttered with defective power plants that further fuel stress and death rather than recovery.
Switching off ATF4 to rescue cells
To test whether ATF4 was driving this spiral, the researchers used small interfering RNA to partially silence ATF4 in cultured corneal endothelial cells. Under the same chronic stress, cells with reduced ATF4 showed lower levels of death‑promoting proteins, healthier mitochondrial membrane potential, less fragmentation, and better survival in viability tests. Importantly, the number of stalled mitophagy structures fell, suggesting that lowering ATF4 helped restore a more effective balance between damage and cleanup. In mice engineered to have only one working copy of the ATF4 gene, UVA exposure caused less activation of a pro‑death partner protein, CHOP, and preserved more normal‑shaped endothelial cells compared with fully ATF4‑sufficient mice.
What this means for people with FECD
For non‑specialists, the message is that one stress messenger, ATF4, can tip corneal endothelial cells from coping to collapsing. When endoplasmic reticulum stress is long‑lasting, ATF4 helps disrupt mitochondria, stalls the cell’s cleanup machinery, and ultimately encourages these vital cells to self‑destruct. Dialing down ATF4—either genetically in mice or with targeted molecular tools in cells—protects mitochondria, improves waste removal, and keeps more cells alive. While this work is still at the laboratory and animal stage, it highlights ATF4 and related stress pathways as promising drug targets that might one day slow or prevent the progression of Fuchs’ dystrophy and reduce the need for corneal transplantation.
Citation: Qureshi, S., Kim, S.Y., Lee, S. et al. ATF4 regulates mitochondrial dysfunction and mitophagy, contributing to corneal endothelial apoptosis. Sci Rep 16, 5960 (2026). https://doi.org/10.1038/s41598-026-36453-x
Keywords: Fuchs endothelial corneal dystrophy, corneal endothelium, mitochondrial stress, mitophagy, ATF4