Clear Sky Science · en
Role of MNX1-mediated histone modifications and PBX gene family in MNX1-induced leukemogenesis
Why a tiny genetic glitch matters for sick children
Most childhood leukemias are now treatable, but a rare form called infant acute myeloid leukemia (AML) remains especially deadly. Many of these infants share the same genetic accident: two chromosomes swap pieces, turning a quiet developmental gene called MNX1 into an overactive driver. This study asks a simple but crucial question: once MNX1 is turned up, what does it actually do inside young blood cells to push them toward cancer—and can that process be interrupted?
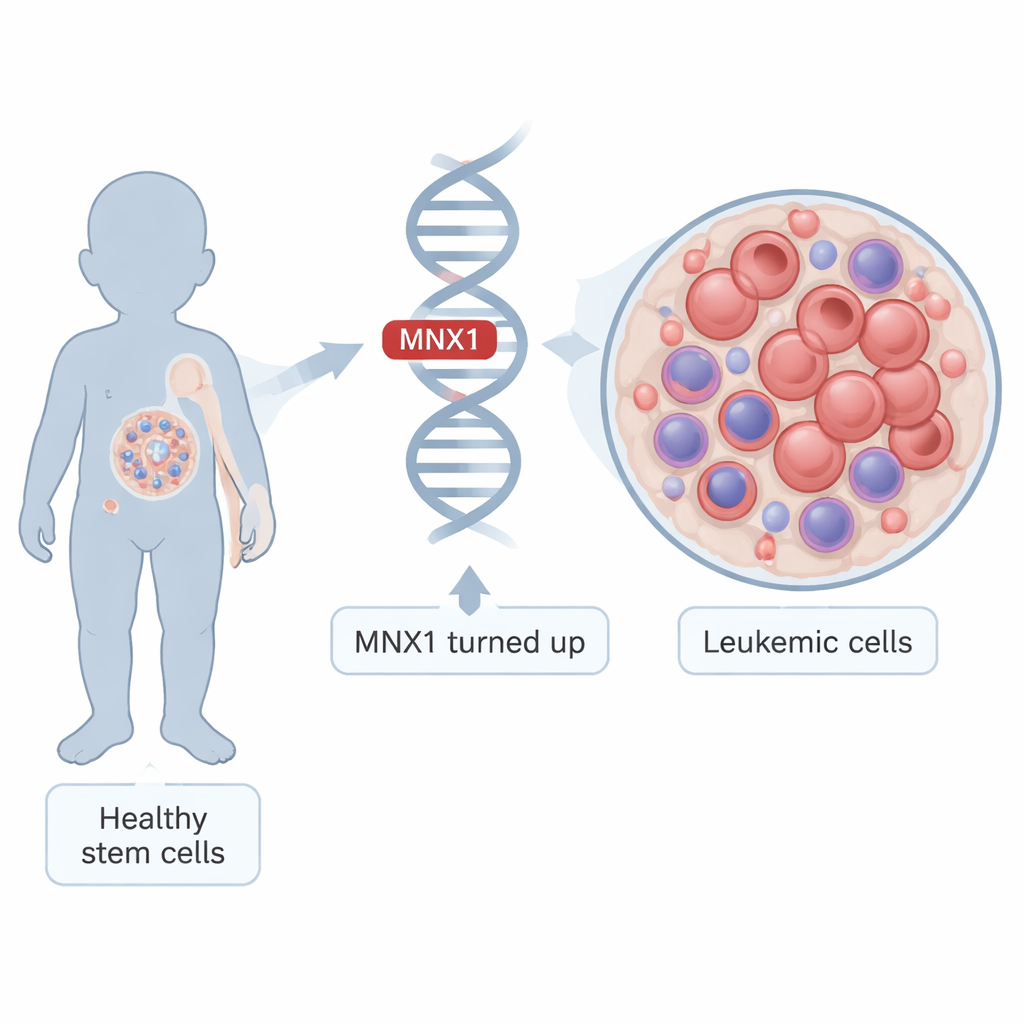
From normal baby blood cells to runaway growth
Leukemia arises when immature blood cells stop maturing and start multiplying out of control. In infants carrying the t(7;12) chromosome swap, MNX1 becomes abnormally active in very early blood-forming cells found in the fetal liver and bone marrow. The researchers built a mouse model that mimics this situation: they switched on human MNX1 in fetal blood stem cells, transplanted them into mice, and watched as the animals progressed from a pre-leukemic state to full-blown leukemia. By comparing cells at these different stages with healthy controls, they could trace how MNX1 rewires the cell’s internal control systems over time.
How MNX1 rewrites the cell’s instruction book
MNX1 is a transcription factor, a protein that sits on DNA and controls which genes are turned on or off. The team combined several powerful methods—mass spectrometry, RNA sequencing, and chromatin profiling—to see which partners MNX1 works with and which genes it changes. They found that MNX1 teams up with enzymes that add chemical marks to histone proteins, the spool-like structures around which DNA is wrapped. In particular, MNX1 boosts an “ON” mark called H3K4me3 and reduces an “OFF” mark called H3K27me3 at certain spots in the genome. These changes loosen the local DNA structure and make it easier for key growth-related genes to switch on.
A hit-and-run push on a critical control gene
Among many affected genes, one stood out: Pbx1, part of the PBX family of DNA-binding proteins long linked to leukemia. The study shows that MNX1 binds directly to the Pbx1 gene’s control region, increasing the ON mark and stripping away the OFF mark there. This jump-starts Pbx1 expression very early, when cells are still only pre-leukemic. Surprisingly, later on—once leukemia is established—MNX1 itself is no longer strongly attached at that site, yet the Pbx1 gene remains switched on and its histone marks stay in the pro-growth configuration. This suggests a “hit-and-run” mechanism: MNX1 briefly visits key regions of chromatin, leaves behind durable epigenetic marks, and then can depart while the altered state continues to fuel disease.
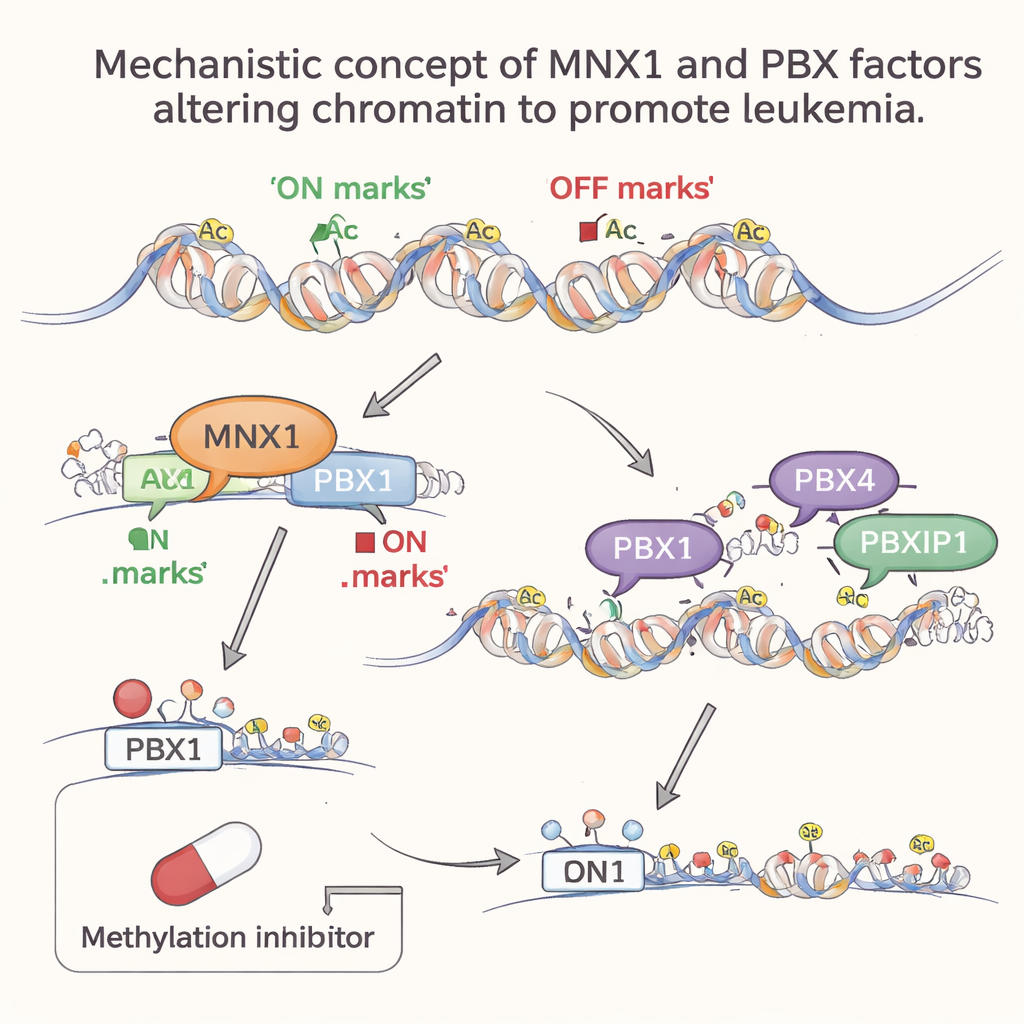
Later helpers join in to lock in the cancer state
As leukemia progresses, other PBX family members, PBX4 and PBXIP1, become more active, but only at this later stage. Genome-wide analyses showed that their preferred DNA motifs are heavily enriched in regions of open, active chromatin that have been remodeled in the wake of MNX1 activity. In other words, MNX1 first reshapes the chromatin landscape and turns on Pbx1; then PBX4 and PBXIP1 move into these newly accessible regions to reinforce abnormal gene programs that promote cell division, block normal blood development, and impair DNA repair. This stepwise involvement hints at a division of labor: PBX1 acts as an MNX1-dependent early switch, while PBX4 and PBXIP1 help maintain the leukemic program.
Blocking the chemical marks that fuel leukemia
Because MNX1 works through enzymes that add methyl groups to histones, the team tested whether a broad methyltransferase inhibitor, Sinefungin, could disrupt this chain of events. In pre-leukemic fetal blood cells expressing MNX1, Sinefungin sharply reduced Pbx1 levels, consistent with blocking the promoter-specific methylation that MNX1 relies on. In contrast, PBX4 and PBXIP1 levels barely changed, fitting with their later, indirect activation during disease progression. Together, these results make a compelling case that infant AML with t(7;12) is driven not just by a broken gene, but by a cascade of durable epigenetic changes that MNX1 sets in motion.
What this means for future treatments
For non-specialists, the take‑home message is that this study maps out a chain reaction: an overactive MNX1 gene rewires the chemical marks on DNA-packaging proteins, switches on PBX1 early, and paves the way for PBX4 and PBXIP1 to help lock cells into a leukemic state. Because these steps depend on specific histone methylation patterns, they offer clear, testable targets for drugs that interfere with those marks. In the long run, therapies aimed at the MNX1–PBX axis or the enzymes that place these epigenetic tags could help turn off the faulty instructions that drive this aggressive leukemia in infants, improving chances for durable cures.
Citation: Malmhäll-Bah, E., Östlund, A., Nilsson, T. et al. Role of MNX1-mediated histone modifications and PBX gene family in MNX1-induced leukemogenesis. Sci Rep 16, 2593 (2026). https://doi.org/10.1038/s41598-026-36367-8
Keywords: infant acute myeloid leukemia, MNX1, PBX1 PBX4 PBXIP1, epigenetic histone methylation, t(7;12) chromosomal translocation