Clear Sky Science · en
Self-supervised learning on graphs predicts non-coding RNA and disease associations
Why hidden RNA matters for our health
Most of us learned that RNA’s main job is to help build proteins. But over the past decade, scientists have discovered vast numbers of “non-coding” RNAs that never become proteins yet still help control how our cells work. Many of these molecules are now known to drive or suppress cancers and other complex diseases. Finding out which non-coding RNAs are linked to which diseases could reveal new ways to diagnose illness early or design more precise treatments—but testing every possibility in the lab would be impossibly slow. This study introduces a powerful computer-based method that can sift through huge biological networks and reliably propose the most promising RNA–disease connections for researchers to check in experiments.
From junk to key cellular players
For years, non-coding RNAs were dismissed as meaningless leftovers of gene activity. We now know that families such as microRNAs, long non-coding RNAs and circular RNAs help orchestrate vital processes, from packaging DNA to switching genes on and off and relaying signals inside cells. Because they sit at so many control points, even small changes in these RNAs can tip the balance toward cancer or other illnesses. Clinicians have already started to see them as potential biomarkers and drug targets. The challenge is scale: there are thousands of different RNAs and hundreds of diseases, and traditional experiments to test each possible link are expensive and time-consuming. That is where computational prediction comes in, offering a way to narrow the search space.
How to read a biological network
Previous computer methods tried to predict RNA–disease links by breaking big data tables into simpler pieces or by training machine-learning models on known examples. These approaches helped, but they often ignored how RNAs and diseases are woven together into networks. Modern “graph neural networks” treat RNAs and diseases as dots connected by lines, much like a social network. They can learn patterns in who is linked to whom. However, most of these graph methods need many reliable training examples and lots of carefully crafted input features. That makes them sensitive to missing data, noisy measurements and overfitting—performing well on known data but failing when asked to predict new associations.
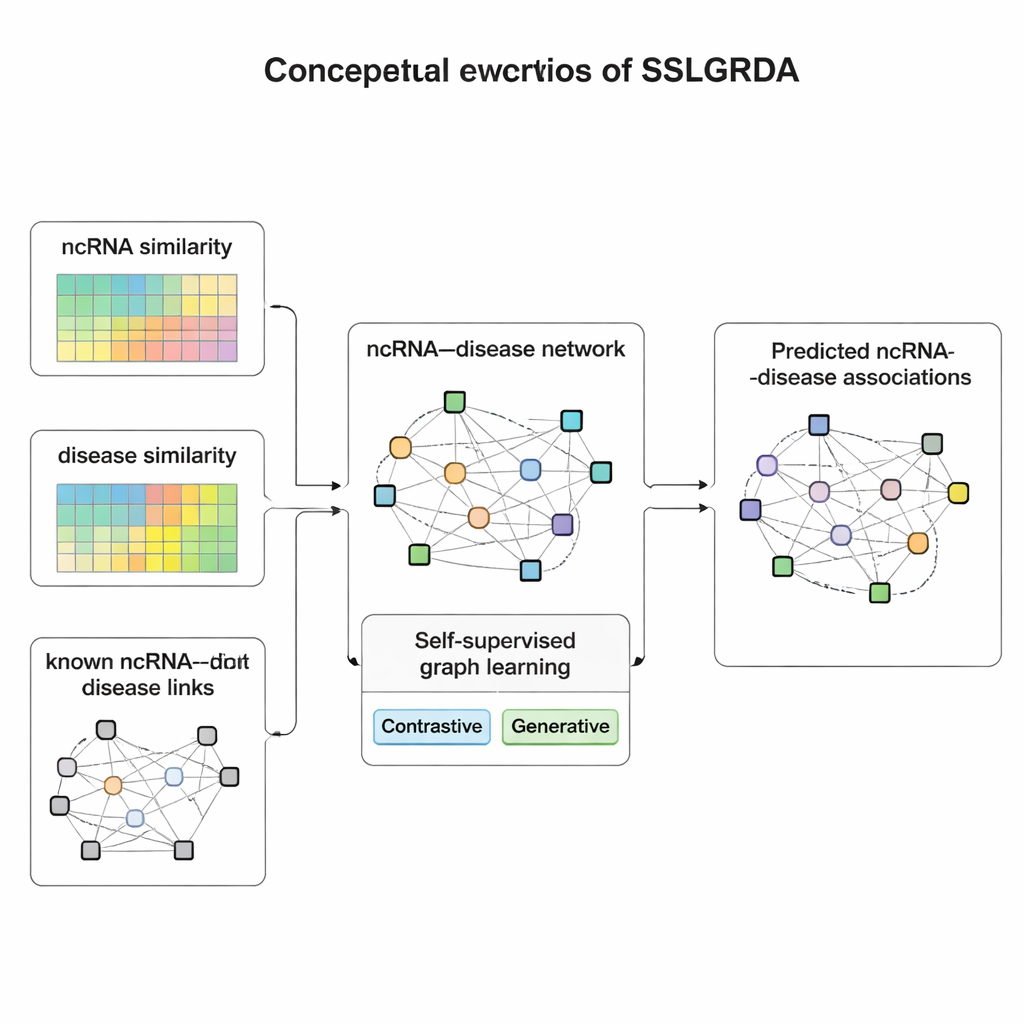
Learning from the data itself
The authors present SSLGRDA, a new framework that teaches a graph model to learn useful patterns without relying heavily on labeled training data. The key idea is “self-supervised learning”: instead of being told which RNA pairs with which disease, the model invents its own practice tasks based solely on the structure and attributes of the network. The researchers build two types of graphs. One keeps RNAs and diseases as different node types connected by known links. The other blends them into a single large network that also includes similarity information—how alike two RNAs or two diseases are—so that even sparsely connected items gain supportive neighbors. On top of these graphs, SSLGRDA uses two styles of self-training. Contrastive strategies ask the model to recognize that different “views” of the same node (for example, its connections versus its attributes) should lead to similar internal representations, while clearly separating unrelated nodes. Generative strategies deliberately hide parts of the input features and challenge the model to reconstruct them, encouraging it to capture deeper structure rather than memorizing noise.
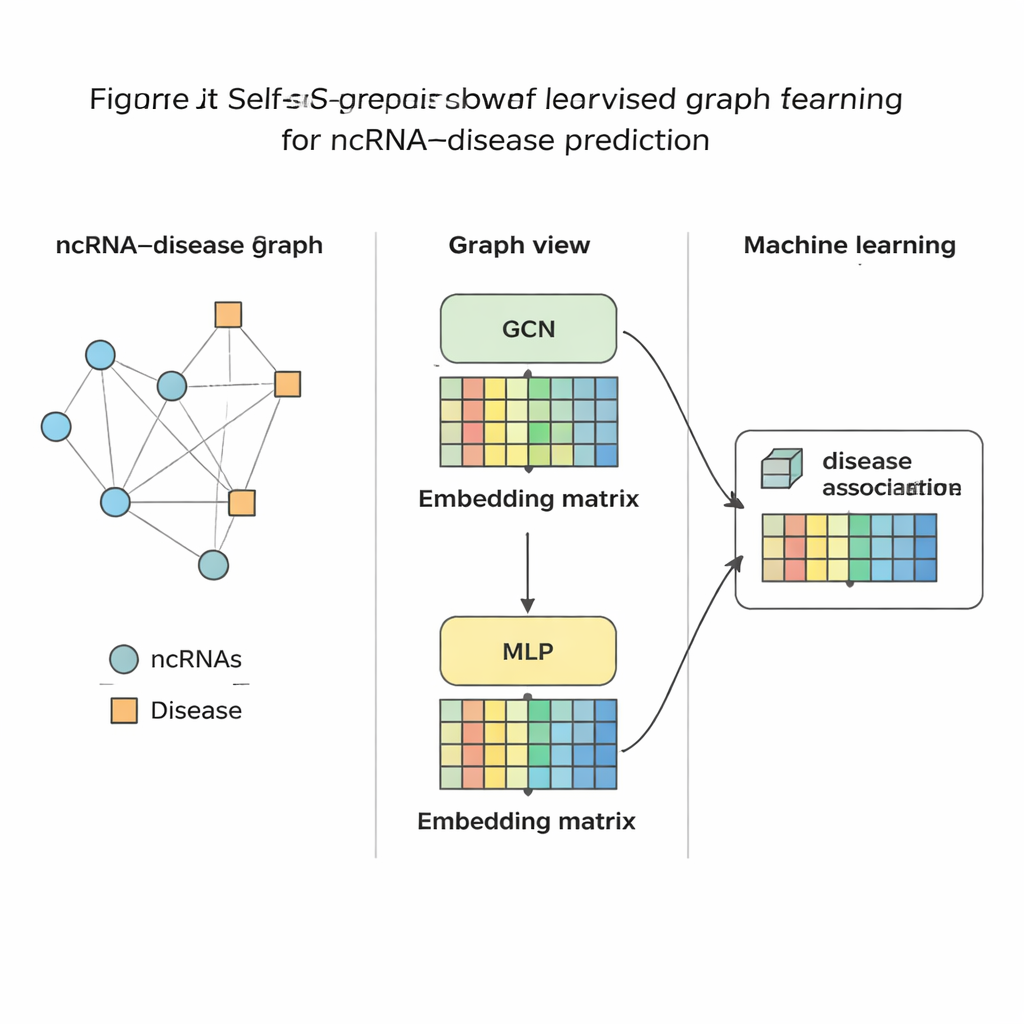
Putting the method to the test
Once SSLGRDA has distilled each RNA and disease into a compact numerical fingerprint, a standard machine-learning classifier is trained to judge whether a link between them is likely or not. The authors evaluated this approach on nine different datasets covering three major RNA types and hundreds of diseases. Across the board, their contrastive self-supervised variants on the blended (homogeneous) graph performed best, beating a range of existing tools, including strong graph-based baselines. The method not only achieved higher accuracy in global tests but also ranked the correct partners near the top when focusing on one RNA or one disease at a time—crucial for real-world use where a biologist might start from a single cancer and ask which RNAs to study. They further showed that the same ideas transfer well to other biomedical networks, such as those connecting microbes to diseases or drugs.
From predictions to potential therapies
To demonstrate practical value, the team applied SSLGRDA to search for new non-coding RNAs involved in breast cancer, colon cancer and several other conditions. Many of the top-ranked suggestions were later confirmed in independent databases or scientific reports, supporting the model’s ability to spot biologically meaningful patterns. For non-specialists, the takeaway is that this work provides a smarter way to mine the ever-growing tangle of biological data for hidden disease clues. By automatically learning how RNAs and diseases cluster and interact, self-supervised graph methods like SSLGRDA can guide laboratory researchers toward the most promising targets, potentially speeding the path from raw data to better diagnostics and treatments.
Citation: Wu, Q., Tang, S. Self-supervised learning on graphs predicts non-coding RNA and disease associations. Sci Rep 16, 5231 (2026). https://doi.org/10.1038/s41598-026-36030-2
Keywords: non-coding RNA, disease association, graph neural networks, self-supervised learning, computational biology