Clear Sky Science · en
Sepsis-associated skeletal muscle wasting is ameliorated by pharmacological inhibition of the STAT3 signaling pathway in mice
Why severe infections can steal your strength
Surviving a life-threatening infection like sepsis is only half the battle. Many patients leave intensive care so weak that walking, climbing stairs, or even lifting their arms becomes a struggle. This study asks a simple but urgent question: can we stop the body from chewing up its own muscles during sepsis, and if so, how? Using mice, muscle cells in a dish, and observations from people in an intensive care unit, the researchers trace a key signaling route that drives muscle loss—and show that a targeted drug can partly block the damage.
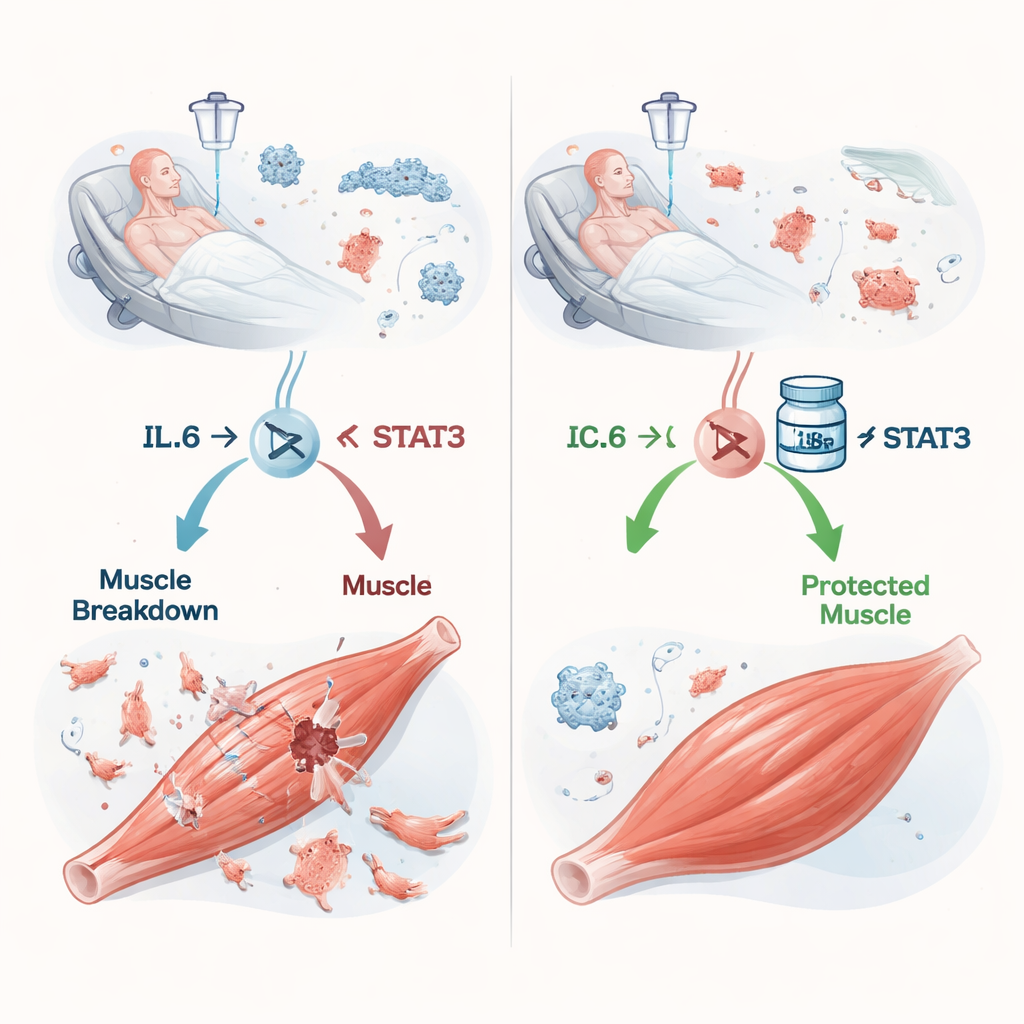
A chain reaction from infection to muscle loss
Sepsis occurs when the body’s response to infection spins out of control, flooding the bloodstream with inflammatory molecules. One of the most important of these is interleukin‑6 (IL‑6). Earlier work hinted that IL‑6 can tell muscles to break down their own proteins, but the details were murky. The authors focused on STAT3, a protein inside cells that relays IL‑6’s message to the nucleus, where genes are switched on or off. In mice given a cecal slurry—essentially a controlled, mixed-bacteria infection—IL‑6 levels in blood and leg muscles shot up as sepsis worsened. At the same time, STAT3 became activated in muscle, and animals lost weight, muscle mass, and grip strength in a severity‑dependent fashion that closely resembled what is seen in critically ill patients.
How sepsis reprograms muscle cells
To understand what sepsis was doing inside muscle fibers, the team analyzed gene activity in the tibialis anterior, a major leg muscle. Thousands of genes changed their activity in septic mice compared with healthy controls. Pathways involved in inflammation, cellular stress, and especially IL‑6/STAT3 signaling were switched on. Two major protein‑disposal systems ramped up: the ubiquitin–proteasome system, which tags specific muscle proteins for destruction, and autophagy, a more general recycling process. Key “muscle shredding” enzymes, MuRF1 and atrogin‑1, increased sharply, while growth‑promoting pathways and classical cell‑death signals remained largely unchanged. In parallel experiments, cultured mouse muscle cells exposed to lipopolysaccharide (LPS), a component of Gram‑negative bacterial walls, showed the same pattern: activation of IL‑6 and STAT3, upsurge of MuRF1 and atrogin‑1, more autophagy, and visible thinning of the muscle fibers.
Blocking a key switch to protect muscle
The central experiment tested whether shutting down STAT3 could spare muscle. Mice with sepsis received a small‑molecule STAT3 inhibitor called C188‑9, starting one hour after infection and then daily. The drug did not soften the initial “cytokine storm”—blood levels of IL‑6 and another inflammatory factor, TNF‑α, stayed high, and body weight and appetite did not quickly recover. Yet C188‑9 clearly protected skeletal muscle: treated mice retained more tibialis muscle mass, gripped more strongly, and had larger muscle fibers under the microscope than untreated septic mice. Inside the muscles, C188‑9 sharply reduced activated STAT3 and lowered MuRF1 and atrogin‑1 levels, but left autophagy markers largely unchanged. In dish experiments, pretreating muscle cells with C188‑9 similarly blunted STAT3 activation and the rise of MuRF1 and atrogin‑1, and prevented LPS‑induced shrinking of the fibers, again without shutting down autophagy.
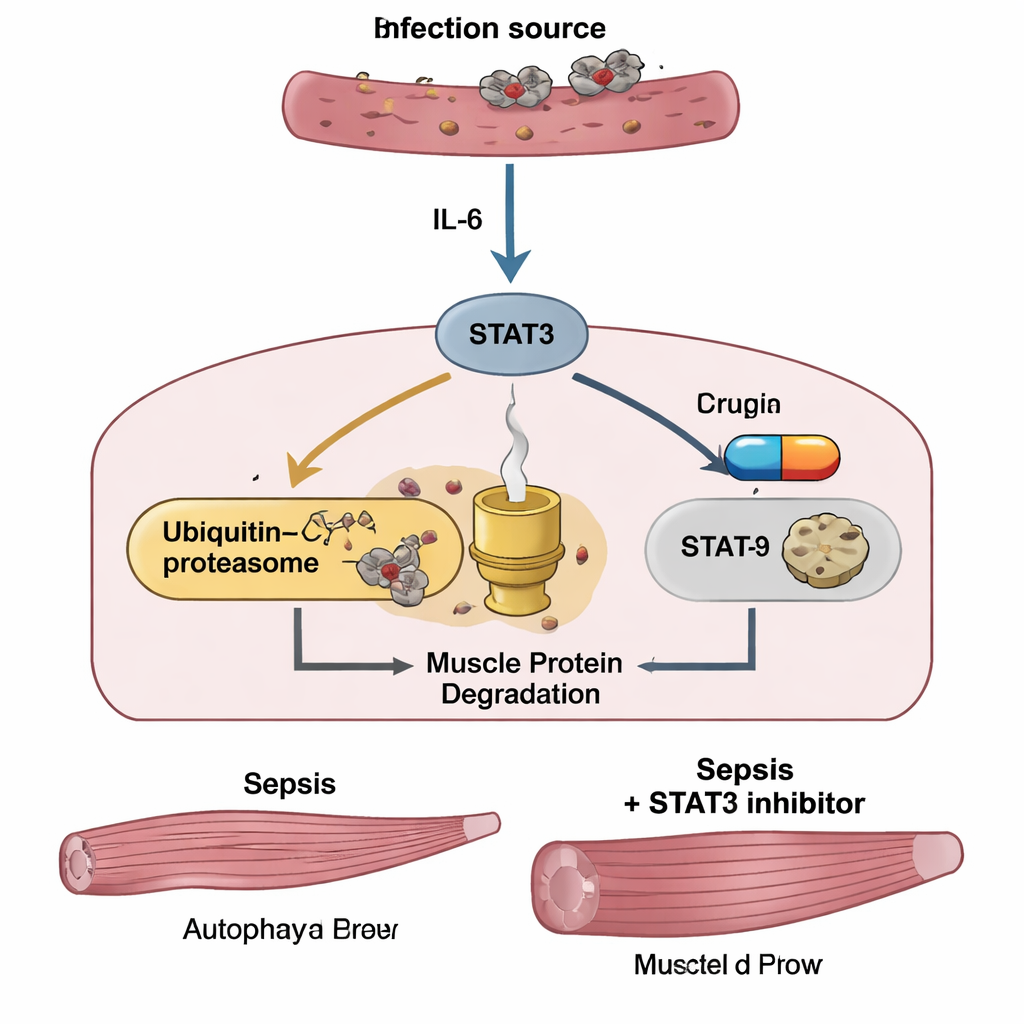
Clues from patients in the intensive care unit
To see whether these mechanisms matter in people, the researchers followed 67 adults with sepsis admitted to an intensive care unit in Japan. Blood tests at admission showed that patients with septic shock had especially high IL‑6 levels. Across the group, IL‑6—but not TNF‑α—tracked closely with sepsis severity scores and with blood markers of inflammation and muscle damage. In a subgroup of 45 patients who underwent two CT scans of the abdomen, IL‑6 levels on admission predicted how much the psoas muscle at the lower spine shrank over the next one to three weeks. Those who lost the most muscle had markedly worse two‑year survival than those who maintained more of their muscle mass, underscoring that sepsis‑related wasting is not just cosmetic—it is linked to long‑term mortality.
What this could mean for future treatments
Put together, the mouse, cell, and human data outline a plausible story: during sepsis, soaring IL‑6 activates STAT3 in muscle, which in turn cranks up a protein‑degrading system that strips muscle fibers of their contractile machinery. Autophagy also increases but appears less directly controlled by STAT3. By pharmacologically blocking STAT3 with C188‑9, the researchers could interrupt this “self‑cannibalization” route in mice and cultured muscle cells, preserving strength even while the infection and inflammation raged on. Although this work is still preclinical and does not prove that STAT3 inhibitors will help human patients, it points to the IL‑6/STAT3 axis as a promising target for drugs aimed at preventing or reducing the profound muscle weakness that haunts many survivors of sepsis.
Citation: Ono, Y., Saito, M., Yoshihara, I. et al. Sepsis-associated skeletal muscle wasting is ameliorated by pharmacological inhibition of the STAT3 signaling pathway in mice. Sci Rep 16, 5008 (2026). https://doi.org/10.1038/s41598-026-35815-9
Keywords: sepsis, muscle wasting, STAT3, inflammation, critical illness recovery