Clear Sky Science · en
Schleyer-type hyperconjugative aromaticity in CH isomers of diazoles revealed by DFT and NBO analysis
Why ring-shaped molecules matter
Chemists have long known that some ring-shaped molecules are unusually stable and behave in special ways. This property, called aromaticity, underpins everything from the smell of gasoline to the function of many medicines. In this study, researchers explored how tiny chemical attachments, or substituents, can dial this stability up or down in a family of nitrogen‑containing rings called diazoles. By understanding and controlling this subtle effect, scientists can design molecules with more predictable reactivity, which is crucial for drug discovery and advanced materials.
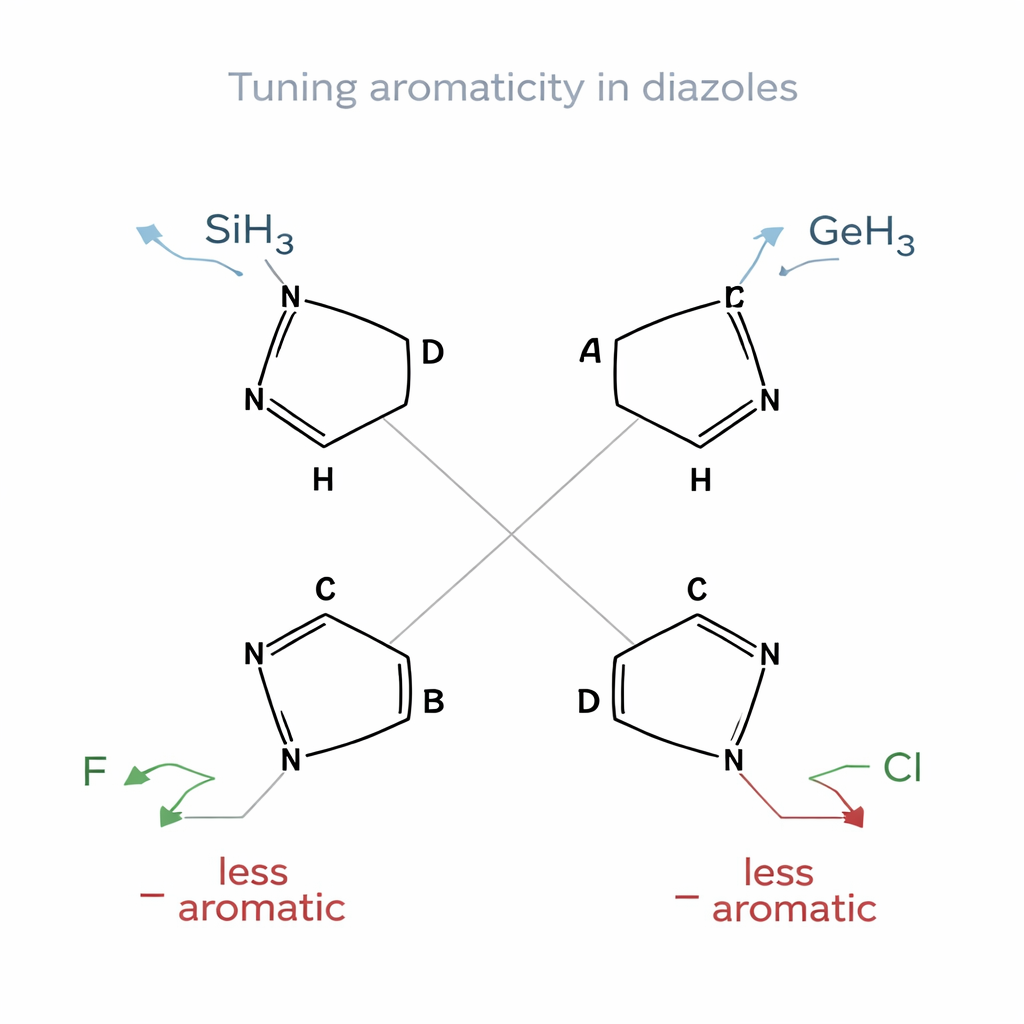
Shaping stability with small changes
Diazoles are five‑atom rings that contain two nitrogens and three carbons; here, the authors focused on less common “CH tautomers,” where one ring position is a carbon–hydrogen unit rather than the usual nitrogen–hydrogen form. They examined four different arrangements of the nitrogen atoms, labeled A through D, each of which changes how electrons are distributed around the ring. Onto these scaffolds they attached a series of simple groups, such as hydrogen, methyl, halogens like fluorine and chlorine, heavier atoms such as silicon and germanium, and classic electron‑donating or withdrawing groups like amino (–NH₂), hydroxy (–OH), cyano (–CN), and boron hydride (–BH₂). Using quantum chemical calculations, they asked how each substituent altered the ring’s aromatic character and overall stability.
How the team measured aromaticity
Aromaticity cannot be observed directly, so the team used several complementary yardsticks. Structural indices, such as HOMED and the Bird Index, track how equal the bond lengths in the ring are; more equal bonds generally signal stronger aromatic character. Magnetic indices, known as NICS values, probe tiny magnetic fields generated by circulating electrons, the hallmark of aromatic rings. Finally, an electronic method called Natural Bond Orbital (NBO) analysis quantifies how strongly electrons can flow from one bond into another, giving a measure of hyperconjugative stabilization. By comparing these different indicators, the researchers built a multidimensional picture of how each substituent affects electron delocalization in the diazole rings.
Winners and losers in boosting ring currents
A clear pattern emerged. Substituents containing silicon (–SiH₃) and germanium (–GeH₃) consistently strengthened aromaticity across all four diazole families. Rings bearing these groups showed more uniform bond lengths, stronger calculated ring currents, and large stabilization energies from electron donation into the ring’s electron system. This behavior fits the concept of Schleyer‑type hyperconjugative aromaticity, where certain σ‑bonds act as powerful donors into the aromatic circuit. A small, strained cyclopropyl‑like bridge (–CH₂–CH₂–) also enhanced aromaticity, acting as an intermediate‑strength donor. In stark contrast, fluorine, and to a lesser extent chlorine, tended to drain electron density from the ring, flattening ring currents and in some cases nearly erasing aromatic character altogether.
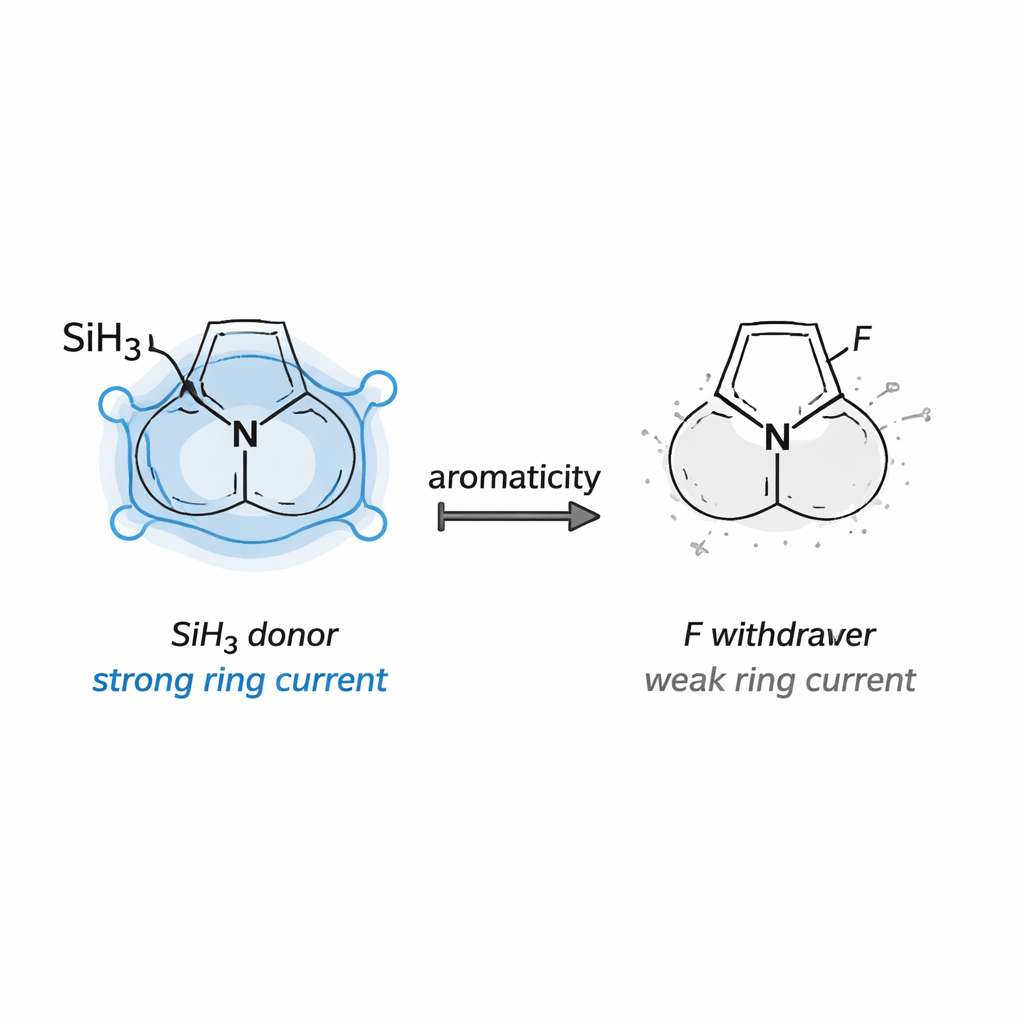
Surprises from classic donors and acceptors
Traditional π‑donor groups such as –NH₂ and –OH, often used to enrich electron density in aromatic systems, had only modest effects here. They slightly improved bond equalization and ring currents but never matched the impact of the silicon and germanium substituents. Even more striking was the contrast between the cyano (–CN) and boron hydride (–BH₂) groups. Both are formally electron‑poor, but they behaved very differently. The cyano group generally weakened aromaticity by pulling electron density away from the ring. By comparison, –BH₂ acted as a surprisingly strong promoter of aromaticity: its bonds donated electron density into the ring framework, much like –SiH₃ and –GeH₃, generating sizeable hyperconjugative stabilization.
One story told by many measurements
When the authors compared all of their indices, the message was consistent. Structural measures, magnetic responses, and electronic stabilization energies all moved in step: rings that looked more benzene‑like geometrically were also those with stronger computed ring currents and greater hyperconjugative stabilization. Fluorinated systems clustered at the low‑aromaticity end of every scale, while silicon, germanium, cyclopropyl‑like, and –BH₂‑substituted rings sat at the high‑aromaticity extreme. For a general reader, the takeaway is that by choosing the right substituents and positions on a ring, chemists can fine‑tune the flow of electrons in tiny molecular circuits. This work maps out how that tuning works in diazole scaffolds, offering practical design rules for crafting more stable, more reactive, or more controllable aromatic molecules.
Citation: Dehkordi, P.N., Saeidian, H., Mirjafary, Z. et al. Schleyer-type hyperconjugative aromaticity in CH isomers of diazoles revealed by DFT and NBO analysis. Sci Rep 16, 7131 (2026). https://doi.org/10.1038/s41598-026-35776-z
Keywords: aromaticity, diazoles, hyperconjugation, substituent effects, computational chemistry