Clear Sky Science · en
A quantitative study of cytotoxic compounds using graph based descriptors and machine learning
Why this research matters for future cancer drugs
Cancer drugs that kill tumor cells, known as cytotoxic drugs, often walk a tightrope between saving lives and causing serious side effects. To design safer and more effective treatments, scientists need fast, reliable ways to predict how these drugs move through the body—how well they are absorbed, how easily they cross cell membranes, and where they end up. This study shows how mathematical descriptions of drug molecules, combined with modern machine learning, can accurately estimate a key property that controls this behavior, potentially speeding up the search for better cancer therapies.
A key surface that controls where drugs can go
One central idea in the paper is the topological polar surface area, or Top_PSA. In simple terms, this is a number that reflects how much of a molecule’s surface is made up of “polar” regions—parts that like water and can form hydrogen bonds. Molecules with very high polar surface areas often struggle to cross fatty cell membranes and may be poorly absorbed when taken by mouth. Molecules with very low polar surface areas can slip through many barriers too easily, sometimes causing unwanted side effects in sensitive tissues like the brain. Top_PSA has become a popular shortcut for estimating these transport properties because it can be computed quickly from a 2D drawing of a molecule, without needing slow 3D simulations.
Turning molecular drawings into numbers
The researchers assembled a curated set of 156 different cytotoxic compounds drawn from real anticancer drugs and experimental agents. They then converted each molecule into 58 so‑called descriptors—numbers that capture features such as how many atoms it has, how many rings, how flexible its bonds are, how many atoms can form hydrogen bonds, and how polar or electronegative different parts are. Many of these descriptors come from graph theory, where a molecule is treated as a network of connected nodes and links. This rich numerical portrait of each molecule served as the input for computer models that try to predict the Top_PSA values calculated by widely used chemistry toolkits.
Testing multiple paths to accurate prediction
To find the best way to link these descriptors to Top_PSA, the team compared several modeling strategies. They tried standard linear regression as well as two “regularized” versions called ridge and LASSO regression, which are designed to cope better with noisy, overlapping information. They also explored different data-preparation schemes: fitting models directly to the raw descriptors, compressing them with principal component analysis (PCA), scaling them in a way that reduces the impact of extreme values (robust scaling), adjusting outliers, and pruning highly correlated features using a measure called the variance inflation factor. Every approach was evaluated carefully using k‑fold cross-validation, a method that repeatedly splits the data into training and test subsets to guard against overfitting.
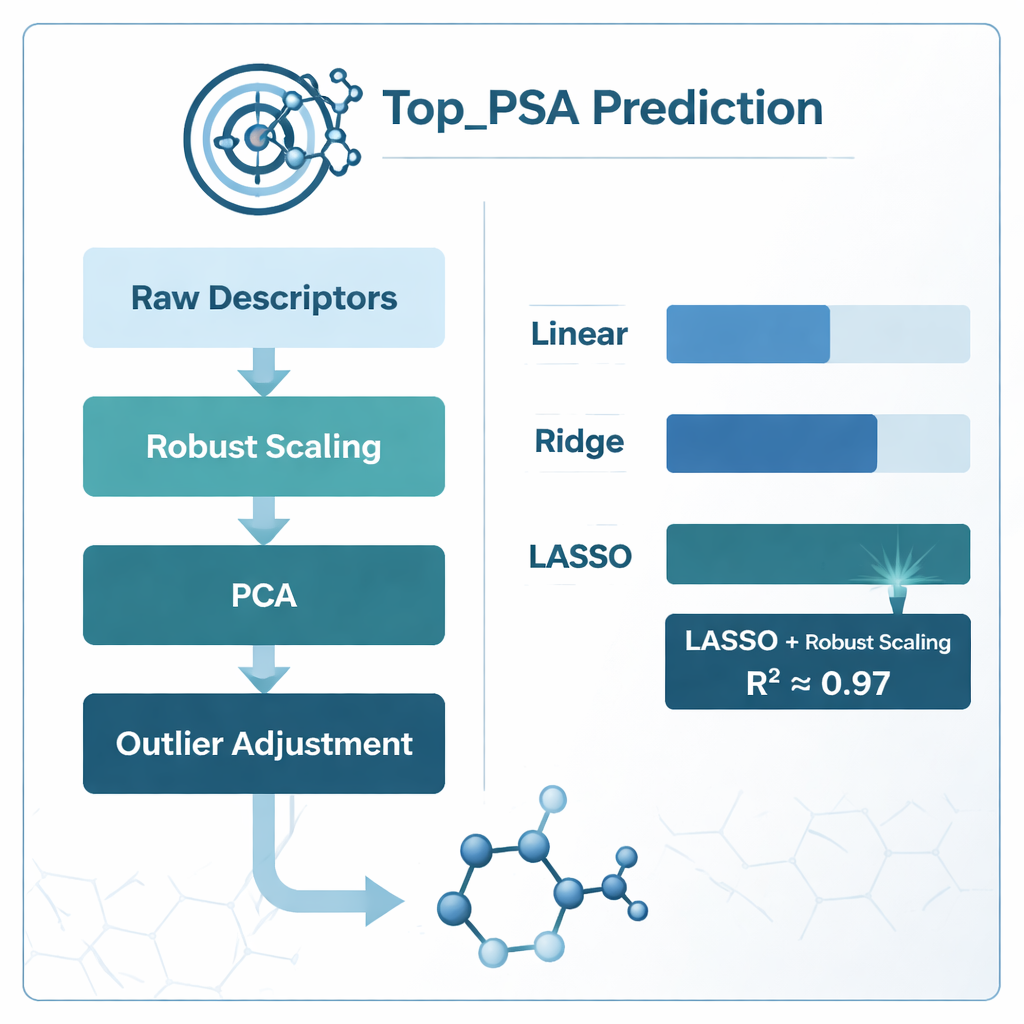
What worked best and what the models learned
The clear winner was the combination of robust scaling with LASSO regression, which achieved a coefficient of determination (R²) of about 0.97—meaning it could explain roughly 97% of the variation in Top_PSA across the 156 drugs. PCA-based models came close in raw accuracy but were harder to interpret chemically because the original descriptors are blended together into abstract components. Simple pruning of correlated descriptors using the variance inflation factor actually hurt performance, suggesting that some overlapping measures still carry useful chemical information. By examining which descriptor weights LASSO kept non-zero, the authors found that the most important factors were the presence of heteroatoms such as nitrogen and oxygen, the ability to give or accept hydrogen bonds, and indices that track how electronegative atoms are arranged across the molecular graph—all features that match the intuitive chemical understanding of polar surface area.
How this can guide better drug design
For readers outside the field, the key message is that carefully prepared mathematical fingerprints of molecules, when paired with well-chosen machine learning methods, can provide fast and reliable estimates of how “sticky” or “slippery” cancer drugs will be as they travel through the body. The study offers practical guidance for other researchers on how to preprocess descriptor data, which modeling approaches to favor, and which shortcuts to avoid. In the long run, such robust, interpretable models of Top_PSA can help chemists filter huge virtual libraries of potential drugs, focusing their efforts on compounds with the right balance of membrane crossing and safety—an important step toward more effective and less toxic cancer treatments.
Citation: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Keywords: cytotoxic drugs, polar surface area, molecular descriptors, machine learning, drug permeability