Clear Sky Science · en
Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism
Why this matters for Alzheimer’s disease
Alzheimer’s disease is often described in terms of sticky protein plaques in the brain, but long before memory fades, the tiny “power plants” inside nerve cells—the mitochondria—and the handling of calcium ions may already be going wrong. This study uses human neurons grown from skin cells of a person carrying a well-known familial Alzheimer’s mutation to ask a simple but crucial question: how early, and in what way, do energy production and calcium balance start to fail?
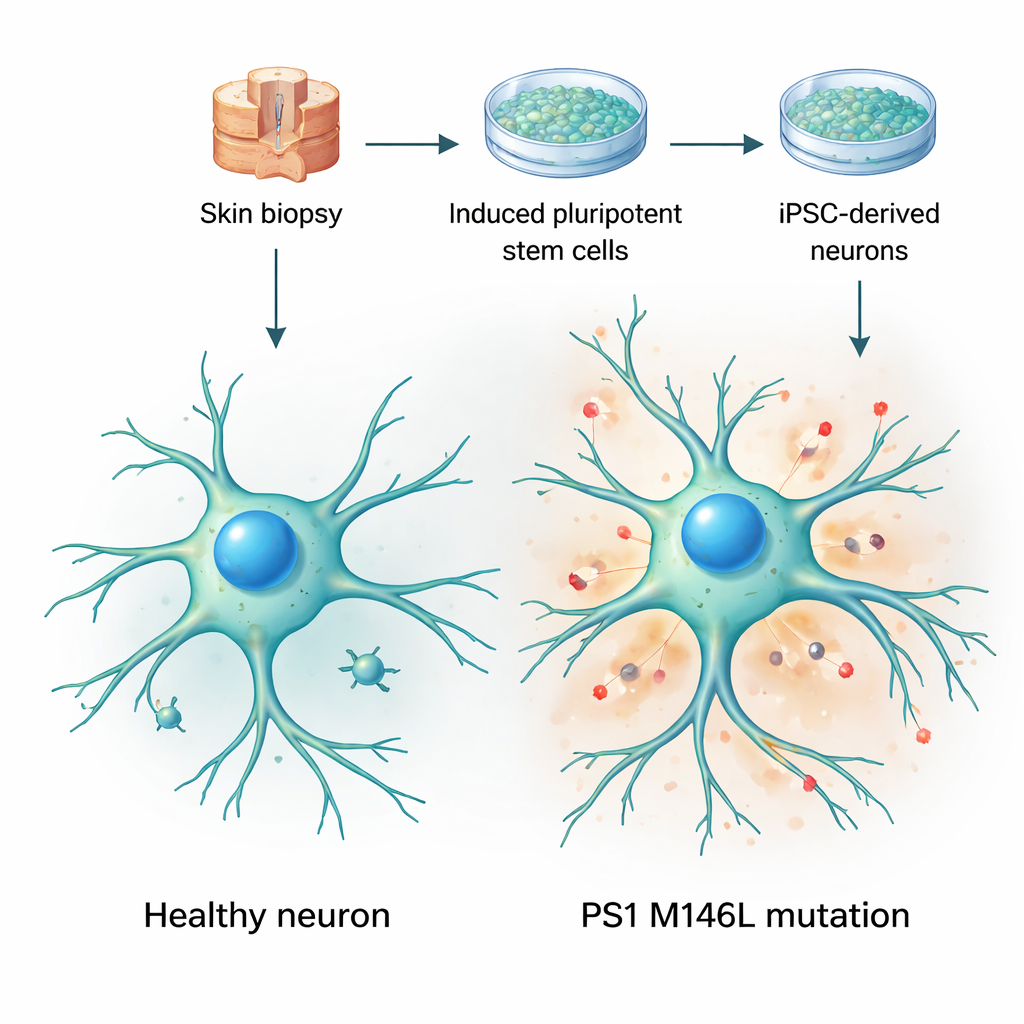
Turning skin cells into living brain models
The researchers began with skin biopsies from two women: one healthy volunteer and one symptom‑free carrier of a presenilin‑1 mutation called M146L, which runs in an Argentine family with early‑onset Alzheimer’s. They reprogrammed the skin cells into induced pluripotent stem cells—cells that can become almost any tissue—and then coaxed them into becoming nerve cells. Over several weeks in culture, these cells acquired typical neuronal shapes, extended long, branched processes, and expressed standard neuronal markers. Importantly, both the control and mutant cells matured at similar rates and looked broadly healthy, which allowed the team to focus on subtle functional changes rather than obvious cell loss or damage.
Electrical signals and calcium under strain
Nerve cells rely on tight control of calcium, a charged atom that acts as a fast on–off switch for many cellular processes. Using fluorescent dyes, the team tracked how calcium levels changed inside the cells when they were electrically stimulated with potassium or activated with signaling molecules. Under simple depolarizing stimulation, neurons carrying the M146L mutation showed weaker calcium increases than control neurons, suggesting problems with maintaining the electrical and ionic gradients that normally drive calcium entry. However, when the researchers triggered a more stressful situation—forcing calcium to leak from internal stores in the endoplasmic reticulum—the difference became clearer. In response to this stress, mitochondria in mutant neurons took up noticeably less calcium than those in control cells, indicating a reduced ability to buffer dangerous calcium surges.
Uncoupling energy use from calcium balance
To understand how this altered calcium handling affects cell metabolism, the investigators measured how much oxygen the neurons consumed—a direct proxy for mitochondrial activity. Surprisingly, neurons with the M146L mutation breathed harder: their basal and maximum oxygen consumption rates, and the amount of oxygen linked to ATP production, were all higher than in control cells. Yet the efficiency of coupling oxygen use to ATP production appeared similar, and there was no increase in mitochondrial number or key ATP‑making enzymes. Instead, mitochondria in mutant neurons were longer and more tubular, with higher levels of a fusion protein called mitofusin‑1, a pattern often seen in cells under chronic, low‑level stress. These hyperactive, elongated mitochondria also generated more reactive oxygen species, unstable molecules that can damage proteins and DNA if not properly controlled.
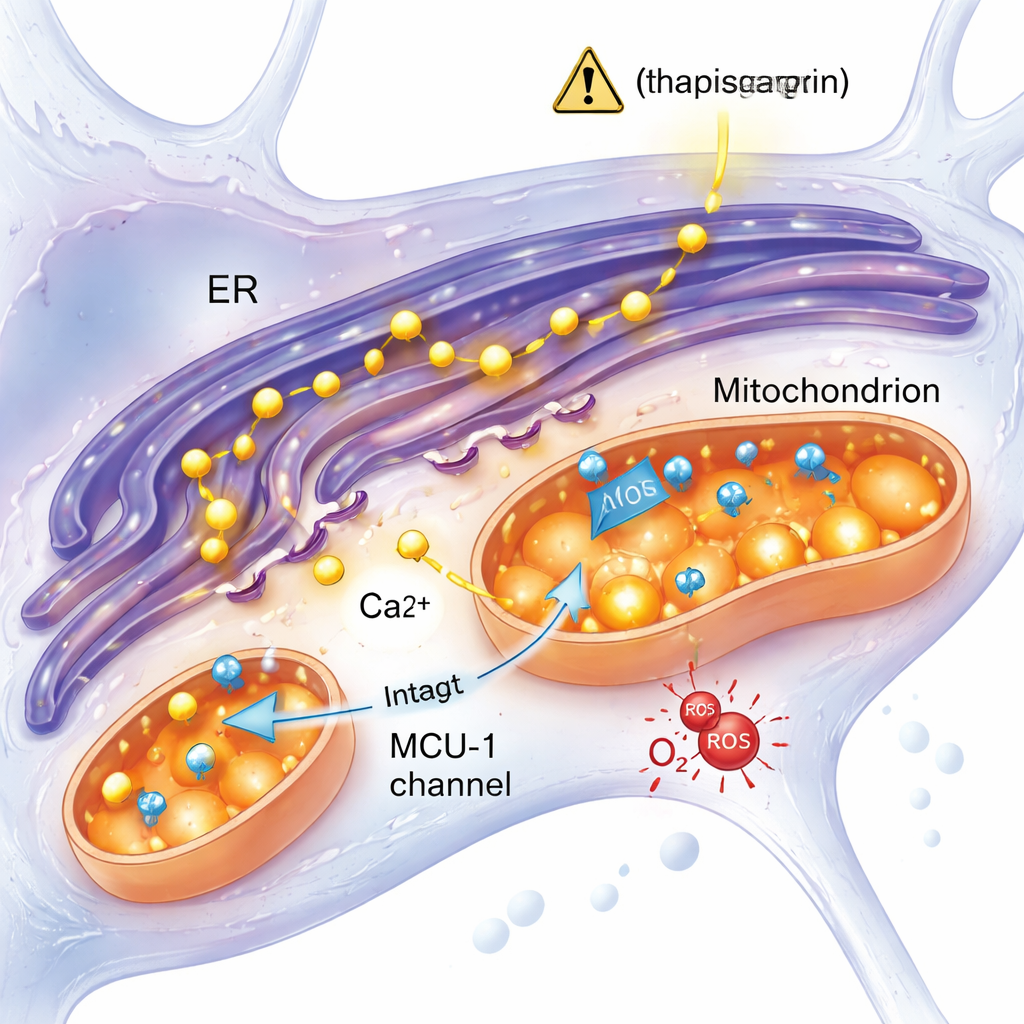
A stress response independent of a key calcium channel
One leading idea in Alzheimer’s research is that excess calcium from the endoplasmic reticulum rushes into mitochondria through a channel called the mitochondrial calcium uniporter (MCU‑1), overloading them and driving dysfunction. This study tested that notion directly. When the team blocked MCU‑1 with a specific inhibitor, both control and mutant neurons showed strong reductions in mitochondrial calcium uptake, confirming that the channel itself worked in both groups. Moreover, when calcium release was triggered through a more physiological pathway involving the IP3 receptor—another key calcium gate—the mutant and control cells responded similarly. These results point away from a broken MCU‑1 channel and instead suggest that the physical and functional contacts between the endoplasmic reticulum and mitochondria, or other aspects of their interaction, are altered in the mutant neurons.
What this means for understanding and treating disease
Taken together, the findings paint a picture of human neurons carrying the PS1 M146L Alzheimer’s mutation as cells that appear normal at rest but react abnormally under stress. Their mitochondria fail to take up enough calcium when internal stores are suddenly released, yet they run hotter—consuming more oxygen and generating more reactive oxygen species—as if locked in a costly compensatory mode. Because this happens in living human‑derived neurons before any clinical symptoms, the work supports the idea that disrupted calcium signaling and early mitochondrial overwork are upstream events in Alzheimer’s, not just late by‑products. For non‑specialists, the key message is that keeping the balance between calcium signals and mitochondrial energy production may be as central to preventing disease as targeting the better‑known amyloid plaques.
Citation: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Keywords: Alzheimer’s disease, mitochondria, calcium signaling, presenilin-1 mutation, iPSC-derived neurons