Clear Sky Science · en
Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression
Why iron inside brain cells matters
Parkinson’s disease is best known for tremors and stiff movements, but deep inside affected brain cells another drama is unfolding: iron, an essential metal, starts to pile up where it should not. This study asks a simple but important question: how does this iron build-up happen, and how might it damage the tiny power plants and recycling centers inside nerve cells? By answering it, the work offers clues to why certain brain regions degenerate in Parkinson’s and related disorders, and points toward new kinds of treatments that go beyond replacing dopamine.
A closer look at a rare genetic clue
The researchers focus on a rare inherited form of Parkinson’s disease, called PARK9, caused by faults in a gene named ATP13A2. This gene makes a protein that sits in lysosomes, the cell’s waste-processing and recycling compartments. People with ATP13A2 mutations can also develop a condition marked by iron deposits in the brain. That link made ATP13A2 an ideal entry point to study how iron balance goes awry. Using a human neuron-like cell line that overproduces the Parkinson’s protein alpha‑synuclein, the team used small RNA fragments to dial down ATP13A2 and then tracked how iron, energy production, and cell health changed.
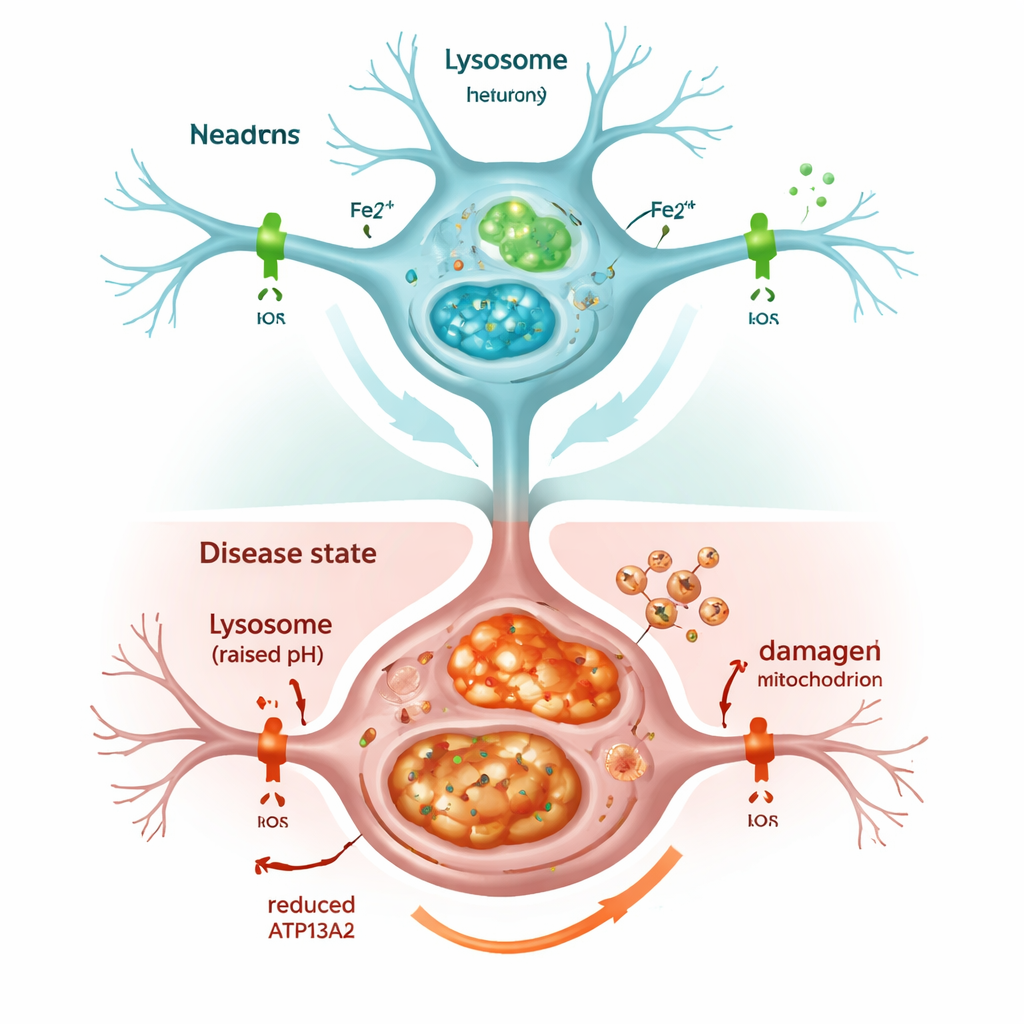
When the cell’s recycling system stalls
Shutting down ATP13A2 quickly weakened lysosomes. Their internal acidity, which is critical for breaking down unwanted material, dropped, and markers of the cell’s cleanup process, known as autophagy, built up instead of being cleared. As a result, alpha‑synuclein accumulated, echoing what is seen in Parkinson’s brains. The cells also showed more iron overall, and especially more of the chemically active form, called Fe2+, inside both lysosomes and mitochondria. The cell responded by making more ferritin, a protein that stores iron, but this was not enough to prevent trouble: the overloaded mitochondria produced excess reactive oxygen molecules, and cell survival declined. Treating the cells with an iron‑binding drug, similar to some used clinically, reduced this oxidative stress and partially rescued cell viability, underscoring that excess iron itself was a key driver of damage.
Iron sensors stop listening to the metal
Ordinarily, cells have a feedback system that notices when iron levels rise and responds by turning down iron import. A protein called IRP2 senses iron, in part through a heme‑dependent signal coming from mitochondria, and then adjusts the production of iron‑carrying proteins on the cell surface. In the ATP13A2‑deficient cells, this safeguard failed. Transporters that bring iron into the cell stayed high even though iron was already elevated. IRP2 protein levels barely changed, and adding extra iron from the outside did not trigger its normal breakdown. The team traced this failure back to the mitochondria: damaged mitochondria breathed less efficiently, showed signs of faulty quality control (mitophagy), and, crucially, lost the ability to make heme, the iron‑containing molecule that helps IRP2 sense iron. Without enough heme, IRP2 did not receive the “too much iron” message and allowed ongoing iron influx.
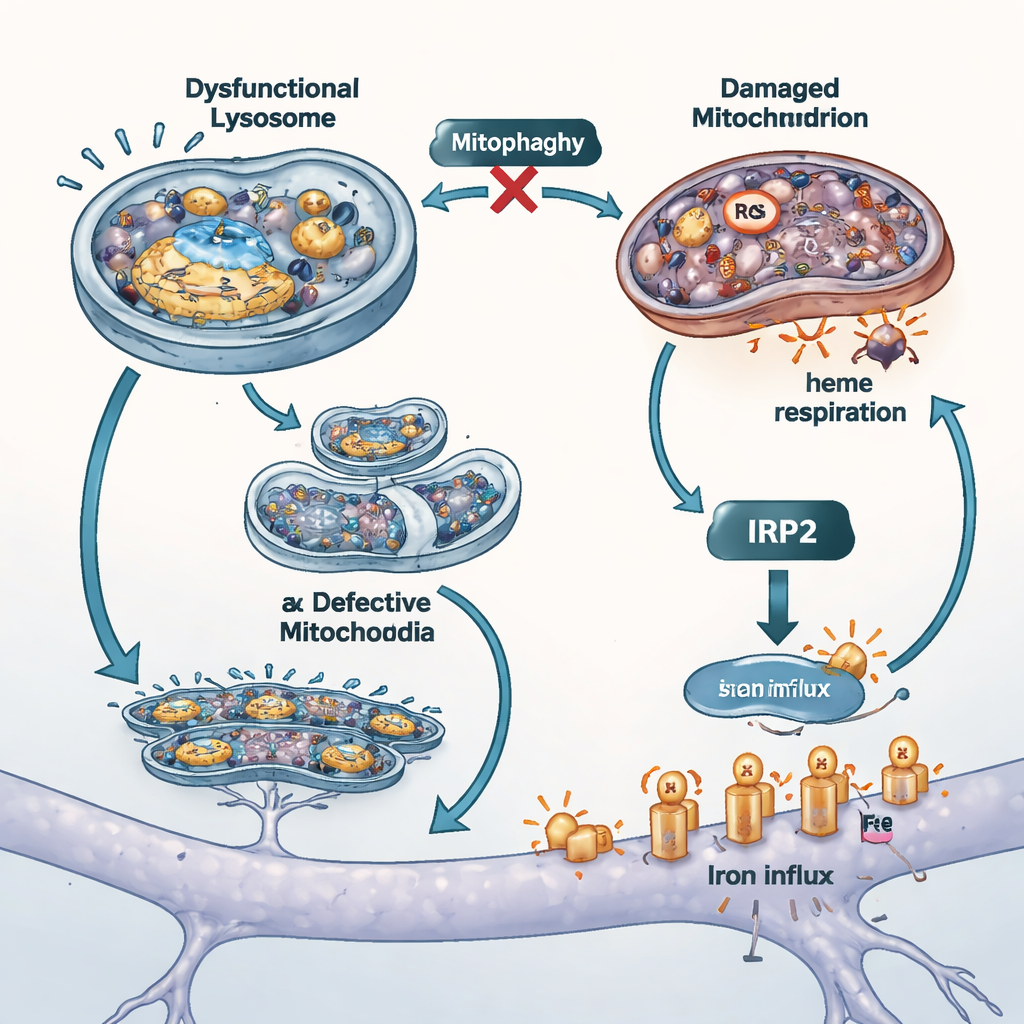
Blocking the iron tap and testing other models
To probe how much this uncontrolled iron entry contributed to cell injury, the scientists blocked two main iron uptake routes. They used an iron‑free version of the blood protein transferrin to compete for one importer, and a small drug to dampen the activity of another transporter called DMT1. Both maneuvers lowered total and free iron inside cells, reduced mitochondrial oxidative stress, and improved survival, suggesting that surface iron channels are important amplifiers of damage when ATP13A2 is lost. The researchers also repeated key experiments in cells lacking another Parkinson’s‑linked gene, PINK1, which is known to impair mitophagy. These cells showed the same combination of iron build‑up and weakened heme production, supporting the idea that mitochondrial quality control and iron balance are tightly intertwined in different forms of the disease.
What this means for Parkinson’s and future treatments
Put simply, the study outlines a vicious cycle. When ATP13A2 is suppressed, lysosomes fail to clear damaged components, including faulty mitochondria. These weakened mitochondria then produce less energy and less heme, blunting the cell’s iron‑sensing system. Iron continues to pour in through surface transporters, collects in vulnerable compartments, and fuels toxic reactions that further injure mitochondria. Over time, this loop may help explain why certain neurons die in Parkinson’s and related iron‑loading brain disorders. The findings suggest that future therapies might not only try to remove excess iron, but also restore proper lysosome function, mitochondrial quality control, and heme production—attacking the problem at its source rather than just mopping up the metal after the fact.
Citation: Murakami, T., Ohuchi, K., Kiuchi, M. et al. Disruption of intracellular iron homeostasis through mitochondrial dysfunction associated with suppression of ATP 13A2 expression. Sci Rep 16, 5007 (2026). https://doi.org/10.1038/s41598-026-35368-x
Keywords: Parkinson’s disease, brain iron, mitochondria, lysosomes, heme synthesis