Clear Sky Science · en
Investigation of the functional hot-spot residues of an enzyme by real-time monitoring of the enzymatic reaction using NMR and computational approaches
Why this matters for future antivirals
Favipiravir is a pill already used against influenza and tested for COVID-19, but it does not fight viruses in the form we swallow. Our own cells must first convert it into an active, virus‑blocking molecule. This study dissects, almost atom by atom, how one human enzyme carries out a crucial activation step, and which tiny parts of the enzyme act as “hot spots” that control how fast and how well the drug is turned on. Understanding these details could guide the design of next‑generation antivirals that are both more potent and more predictable in patients.
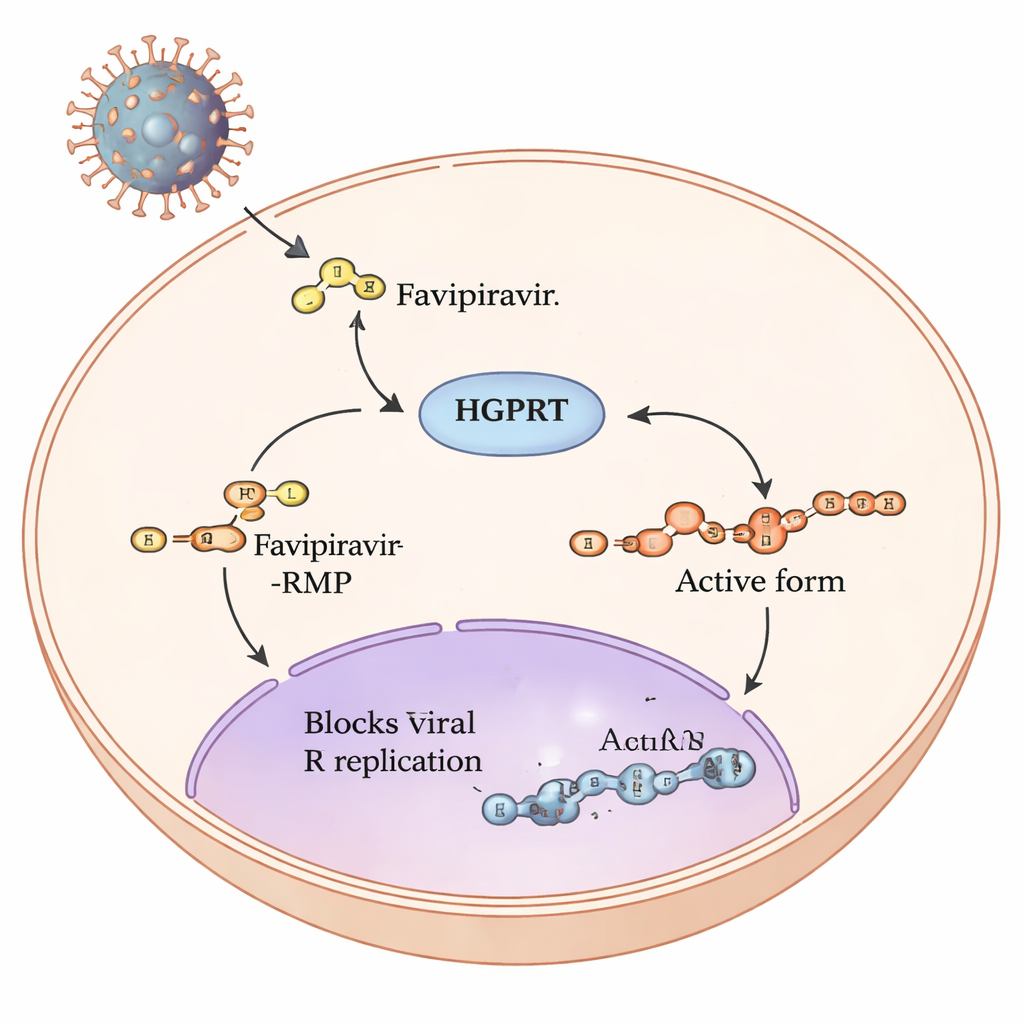
The journey of a prodrug inside our cells
Favipiravir is a so‑called prodrug: once it enters human cells, a series of chemical steps remodels it into a form that can jam the copying machinery of RNA viruses such as influenza and SARS‑CoV‑2. The first and slowest step in this pathway is carried out by a human enzyme called hypoxanthine‑guanine phosphoribosyltransferase, or HGPRT. HGPRT adds a small, sugar‑phosphate group to favipiravir, producing favipiravir‑RMP. Only after this step can other enzymes build the fully active triphosphate form that directly interferes with the viral RNA polymerase. Because this first HGPRT‑driven step acts like a bottleneck on how much active drug is made, the authors set out to pinpoint which parts of HGPRT are most important for handling favipiravir.
Watching chemistry in real time with NMR
Uniquely, favipiravir contains a fluorine atom that behaves like a tiny radio transmitter in a magnetic field. The team exploited this by using fluorine‑19 nuclear magnetic resonance (NMR) spectroscopy to watch, in real time, how much favipiravir and how much favipiravir‑RMP were present in a test tube as the reaction progressed. Because only the drug carries fluorine, the NMR signals are clean and easy to track. By repeatedly recording spectra over 12 hours, the researchers could follow the disappearance of the starting drug and the rise of the modified product, then extract standard kinetic measures such as how fast the reaction proceeds and how tightly the enzyme seems to bind the drug.
Tuning key positions in the enzyme
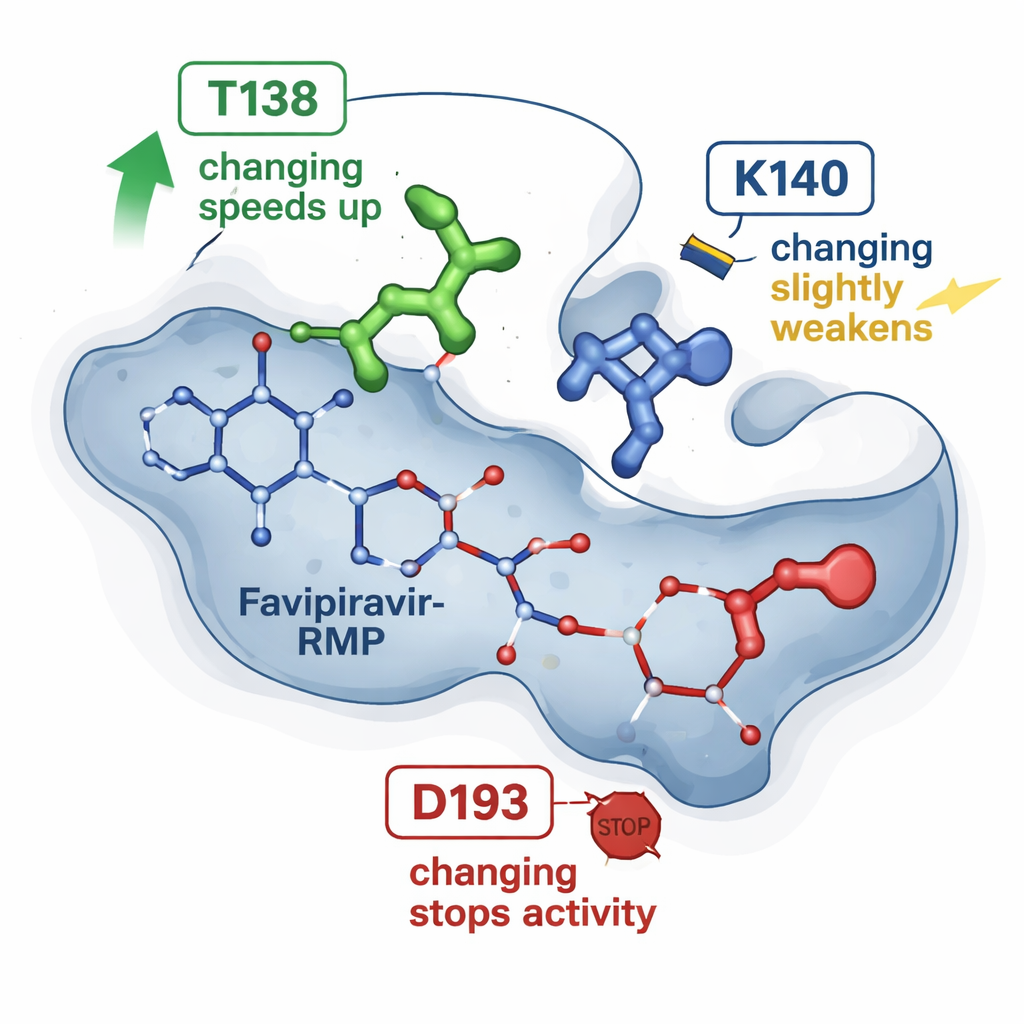
Earlier X‑ray snapshots of HGPRT bound to favipiravir‑RMP had suggested a handful of amino acids that cradle the drug in a pocket. The new work tests three of these positions by making precise single‑letter changes in the protein and comparing each mutant enzyme with the natural one. One change, dubbed T138A, unexpectedly made the enzyme about four to six times faster at converting favipiravir, even though it removed a chemical group that had been thought to help hold the drug. A second change, K140M, modestly slowed the reaction and slightly weakened apparent binding. A third change, D193N, completely abolished the enzyme’s ability to make favipiravir‑RMP, even though the altered protein could still be produced and bind the product. Together, these results show that not all contact points are equal: some act as subtle speed regulators, while others are essential switches.
Simulating the moving parts on a computer
To see beyond static structures, the researchers turned to molecular simulations. Starting from the known three‑dimensional structure of HGPRT with favipiravir‑RMP, they used established computational tools to estimate how strongly the drug binds in each mutant and to run many short molecular dynamics simulations. These simulations track how the atoms jiggle and interact over tens of nanoseconds. The calculations agreed with the NMR‑derived trends: the T138A variant tended to hold favipiravir‑RMP more favorably yet also showed episodes where the drug moved toward an “escape” pathway, guided by another residue (K140) that briefly anchors the phosphate group before release. In contrast, the D193N variant still gripped the product, but likely failed at an earlier catalytic step that requires a magnesium ion, explaining why it lost activity despite stable binding.
A roadmap for smarter antiviral design
By combining real‑time NMR measurements with detailed computer models, this study maps the functional hot spots in HGPRT that govern how efficiently favipiravir is activated. For non‑specialists, the takeaway is that our own enzymes can strongly influence how much active antiviral drug builds up inside cells, and that tweaking either the drug’s shape or the enzyme’s pocket can dramatically change that outcome. The authors’ hybrid strategy offers a general blueprint for probing how other medicines interact with their target proteins, potentially speeding the development of new antiviral compounds that are better matched to the body’s activation machinery.
Citation: Sugiki, T., Yoshida, T., Tsukamoto, M. et al. Investigation of the functional hot-spot residues of an enzyme by real-time monitoring of the enzymatic reaction using NMR and computational approaches. Sci Rep 16, 5896 (2026). https://doi.org/10.1038/s41598-026-35354-3
Keywords: favipiravir, antiviral activation, HGPRT enzyme, NMR spectroscopy, drug design