Clear Sky Science · en
Assessment of the biochemical basis underlying the resistance against systemic amyloidosis
When Tiny Protein Changes Block a Deadly Build-Up
Many chronic inflammatory diseases, from rheumatoid arthritis to tuberculosis, can trigger a rare but often fatal complication called systemic AA amyloidosis. In this condition, a normal blood protein piles up as stiff fibers that clog organs. This study asks a surprisingly hopeful question: can small, natural changes in that protein make some animals largely immune to the disease—and if so, how?
The Hidden Threat of Protein Pile-Ups
AA amyloidosis begins with an inflammatory alarm signal in the blood called serum amyloid A (SAA). During severe or long-lasting inflammation, SAA levels can shoot up thousands of times above normal. In some people and animals, part of this protein misfolds and stacks into long fibers, known as amyloid fibrils, that spread through organs like the spleen and kidneys. Over time these fibers compromise organ function. Yet not every individual with high SAA levels gets amyloidosis, and certain mouse strains remain surprisingly resistant even when pushed toward disease in the lab. Understanding why could point to new strategies to prevent amyloid build-up in humans.
Resistant Mice and Their Special Protein Versions
In mice, most AA amyloid fibrils arise from one version of SAA called SAA1.1, which is strongly linked to disease. However, some mouse strains mainly produce slightly altered versions, named SAA1.5 and SAA2.2, and these strains rarely develop systemic AA amyloidosis. The proteins differ only by a handful of building blocks (amino acids), yet those changes cluster in a tightly packed region that forms the inner core of the disease-causing fibers. The researchers proposed that these small differences do not stop the proteins from clumping altogether, but instead prevent them from adopting the very specific fiber shape that is harmful.
Putting the Proteins to the Test in the Lab
To probe this idea, the team produced all three mouse SAA variants in bacteria and watched how they behaved in test-tube experiments. They monitored fiber formation using a fluorescent dye that lights up when amyloid forms, and verified structures with electron microscopes. The disease-linked SAA1.1 readily formed long, straight fibers. SAA2.2 could also form fibers, but they were thicker, more twisted, and structurally more varied, and they did not trigger the same strong dye signal. SAA1.5, in contrast, failed to form fibers under the conditions tested. When the scientists added tiny samples of real disease fibers taken from sick mouse spleens as “seeds,” SAA1.1 quickly grew new fibers that closely copied the structure of the original, just like a prion. Strikingly, SAA1.5 and SAA2.2 did not grow onto these seeds at all; the ex vivo fibers could not recruit them into the pathogenic shape.
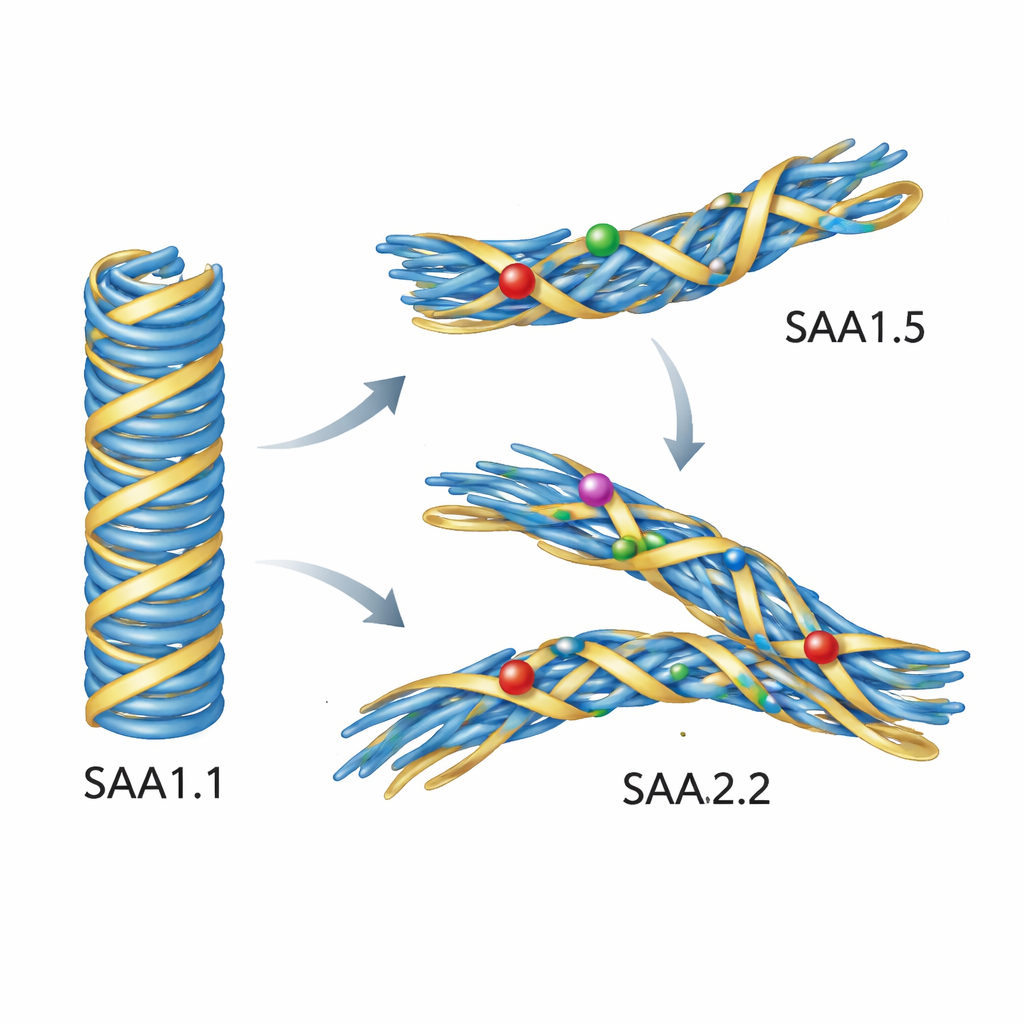
Simulations Reveal Why Mutant Proteins Refuse the Bad Shape
Experiments alone could not show exactly what goes wrong at the atomic level, so the authors turned to detailed computer simulations. They started from a high-resolution structure of a pathogenic mouse AA fibril built from SAA1.1, then computationally substituted in the SAA1.5 and SAA2.2 sequences. When they simulated these fibers in water at body temperature, the SAA1.1-based model stayed remarkably stable. In contrast, fibers built from SAA1.5 or SAA2.2 shifted and distorted. A key loop region in the core moved outward and loosened its contact with the protein’s beginning segment, and several side chains flipped into new orientations. These subtle rearrangements disrupted the tight packing that defines the disease-associated fold. In other words, the variant sequences did not mind forming fibers in general—but they could not comfortably fit the blueprint of the pathogenic AA fibril.
How Nature’s Design Hints at Future Therapies
Taken together, the work shows that “amyloid-resistant” mouse strains are not protected because their SAA cannot aggregate at all. Rather, their versions of SAA are structurally incompatible with the one particular fiber shape that causes systemic AA amyloidosis. The proteins can still clump, but they do so in alternative, apparently benign forms. Similar protective mutations are known in other protein misfolding diseases, including some prion and Alzheimer’s cases. This suggests a broader principle: tweaking a disease-prone protein so that it cannot adopt its toxic architecture—while still letting it function normally—may be enough to prevent illness. In the long run, therapies inspired by these natural “resistant” variants, or by short fragments derived from them, could help steer proteins away from harmful folds and toward harmless ones.
Citation: Moderer, T., Schnell, A.F., Scheurmann, N.J. et al. Assessment of the biochemical basis underlying the resistance against systemic amyloidosis. Sci Rep 16, 1313 (2026). https://doi.org/10.1038/s41598-026-35297-9
Keywords: AA amyloidosis, serum amyloid A, protein misfolding, amyloid resistance, mouse models