Clear Sky Science · en
Stochastic LASSO for extremely high-dimensional genomic data
Finding the Needles in Genomic Haystacks
Modern biology can measure tens of thousands of genes at once, but patient studies often include only a few hundred people. Hidden in this imbalance are small sets of genes that truly matter for predicting disease risk or survival. This paper introduces “Stochastic LASSO,” a statistical method designed to reliably uncover those key genes from oceans of noisy genomic data, even when there are far more genes than patients.
Why Picking the Right Genes Is So Hard
Researchers often rely on tools like LASSO, which shrink unimportant gene effects toward zero while keeping the most informative ones. Classic versions of LASSO, however, struggle when the number of genes dwarfs the number of samples, as is common in cancer genomics. Standard LASSO can only select at most as many genes as there are patients, and it tends to overlook genes that behave similarly to one another. Earlier improvements that add extra penalties can handle some of this correlation, but they may also blur biological meaning by forcing related genes to act as if they all push outcomes in the same direction.
Building Cleaner Random Samples
One promising workaround is to repeatedly fit LASSO on many smaller, randomly drawn subsets of genes, then combine the results. Yet these “bootstrap” approaches still suffer from three problems: correlated genes can cancel each other out, many genes are rarely or never sampled, and pure randomness makes the final selection unstable. Stochastic LASSO tackles these issues head‑on with a new sampling scheme called correlation‑based bootstrapping. Instead of picking genes at random, it deliberately favors genes that are less correlated with those already chosen, yielding smaller sets of genes that are much more independent. It also makes sure every gene is used the same number of times across the bootstrap runs, so no gene is unfairly ignored. 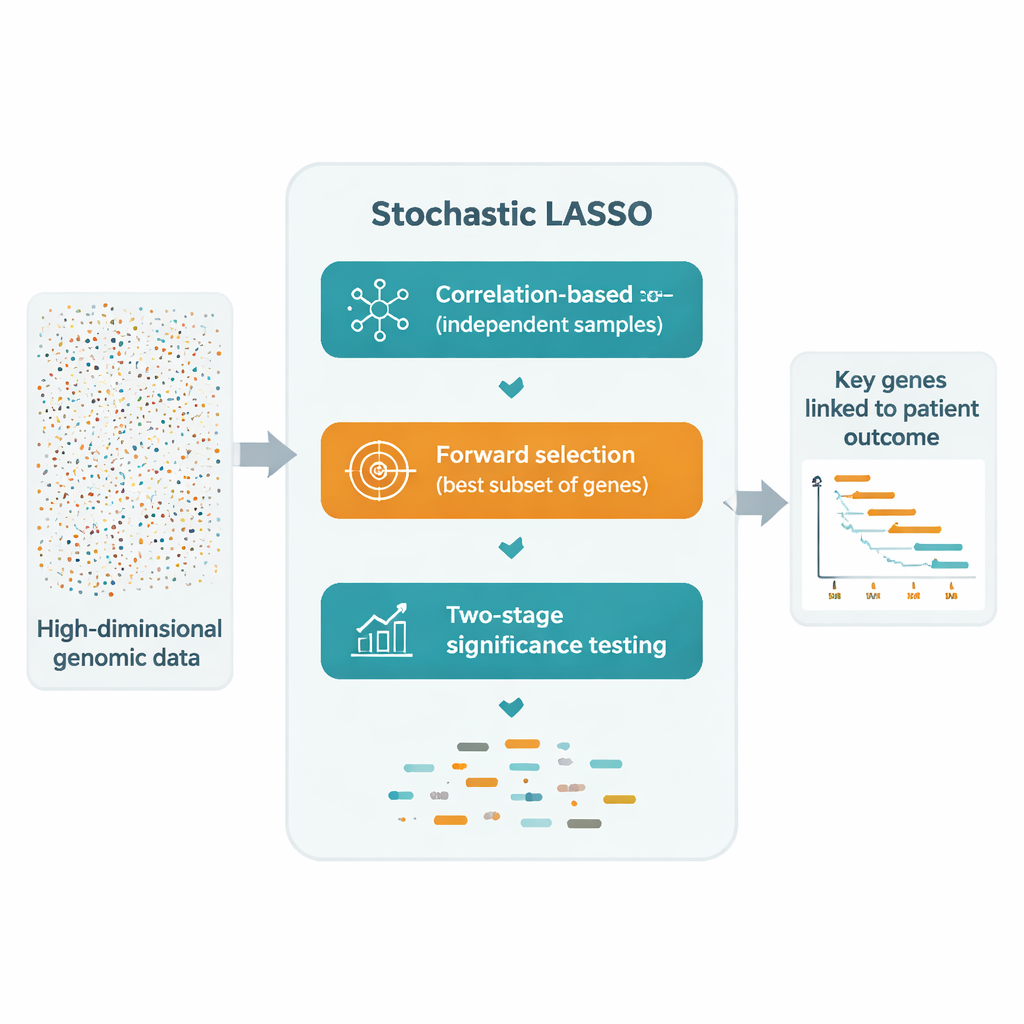
From Local Clues to a Global Gene Set
After building these cleaner subsets, Stochastic LASSO records how large each gene’s coefficient is across all the bootstrap fits. This average absolute effect becomes a “local score” that reflects how consistently important the gene appears. Rather than exhaustively testing every possible combination, the method builds candidate models by adding genes in order of their local scores and evaluates how well each candidate predicts outcomes on separate validation data. In this way, it settles on a compact set of genes whose combined signals best explain the data, while using far fewer trials than traditional stepwise methods.
Testing Which Genes Truly Matter
To move from “often selected” to “statistically convincing,” the authors introduce a two‑stage t‑test. First, they check whether each gene’s average coefficient across bootstraps is clearly different from zero, flagging it as potentially meaningful. Then, among these candidates, they ask whether each gene’s effect is larger than the typical effect size of all candidates. Only genes that pass both tests are declared significant. Because these tests rely on the many bootstrap estimates, Stochastic LASSO can confidently identify more significant genes than there are patients—something conventional LASSO cannot do. 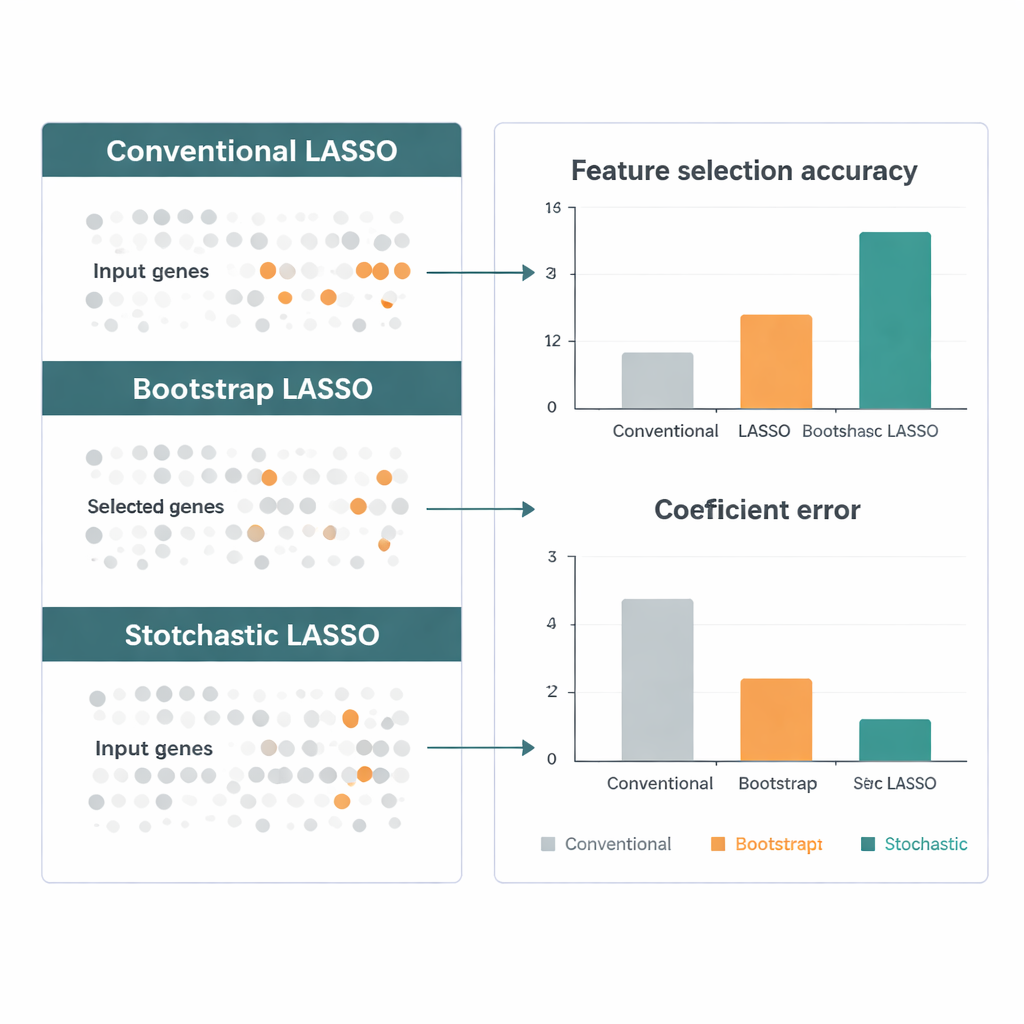
Proving Its Worth in Simulations and Cancer Data
The authors benchmark Stochastic LASSO against several leading LASSO variants using simulated data designed to mimic real genomic studies: very many genes, strong correlations, and known “true” signals. Across multiple scenarios, the new method finds the correct genes more often, estimates their effects more accurately, and remains stable from run to run. They then turn to gene expression data from The Cancer Genome Atlas for brain tumors, including aggressive glioblastoma. Stochastic LASSO highlights hundreds of genes whose activity relates to patient survival and flags biological pathways—such as signaling and drug‑metabolism routes—that have independent support in the literature, suggesting that the method is not just statistically sharper but biologically sensible.
What This Means for Patients and Researchers
For non‑specialists, the key message is that Stochastic LASSO is a smarter filter for genomic big data. It helps scientists separate genuine disease‑related genes from statistical noise, even when data are limited and genes are highly interlinked. By delivering more accurate and more stable gene lists and effect estimates, it can sharpen the search for biomarkers, drug targets, and prognostic signatures in cancer and other complex diseases. Although demonstrated on linear regression, the same framework can be woven into survival models and classification problems, broadening its potential impact across biomedical research.
Citation: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Keywords: genomic feature selection, high-dimensional data, LASSO methods, cancer gene expression, biomarker discovery