Clear Sky Science · en
Loss of Snhg5 disrupts cell-cycle regulation without altering cystogenesis in a mouse model of polycystic kidney disease
Why a kidney gene with a strange name matters
Polycystic kidney disease (PKD) is a common inherited disorder in which countless fluid-filled sacs slowly overrun the kidneys, often leading to kidney failure. Scientists know that faults in two genes, PKD1 and PKD2, drive the disease, but many other genetic players may influence how fast it progresses. This study focuses on an unusual type of gene called a long non-coding RNA, named Snhg5, asking a simple but important question: does this molecule meaningfully shape how PKD unfolds, or is it mostly a bystander?
A closer look at an obscure genetic messenger
Unlike typical genes that provide blueprints for proteins, long non-coding RNAs act more like molecular organizers or switches inside cells. Earlier work showed that Snhg5 is strongly increased in mouse models of PKD and has been linked to several cancers and kidney injuries, hinting it might drive harmful growth. The researchers first mapped where and when Snhg5 is active in normal mice. They found that it is switched on in many organs, with especially high levels in the intestine, and that its activity in the kidney drops sharply after birth, as the organ shifts from rapid growth to a more stable, mature state. Within kidney cells, nearly all Snhg5 molecules reside in the nucleus, the command center that houses DNA, suggesting that Snhg5 helps control gene activity rather than building proteins directly.
Patterns in sick kidneys do not tell the whole story
The team then compared Snhg5 levels in a range of mouse models of PKD. In fast-moving models, where cysts form quickly, Snhg5 was boosted two- to threefold in diseased kidneys, and imaging at the single-cell level showed strong nuclear signals in cyst-lining cells and nearby tissue. Surprisingly, when they looked at a slower, milder mouse model that better mimics the long course of human PKD, Snhg5 did not increase. Even more striking, in kidney tissue from people with advanced autosomal dominant PKD, the human counterpart of the gene, called SNHG5, was actually reduced by more than 90 percent. Together, these results suggest that changes in this RNA accompany cyst formation, but the direction and timing of the change differ between species and disease stages, raising doubts that Snhg5 alone is a straightforward driver of cyst growth.
What happens when the gene is removed
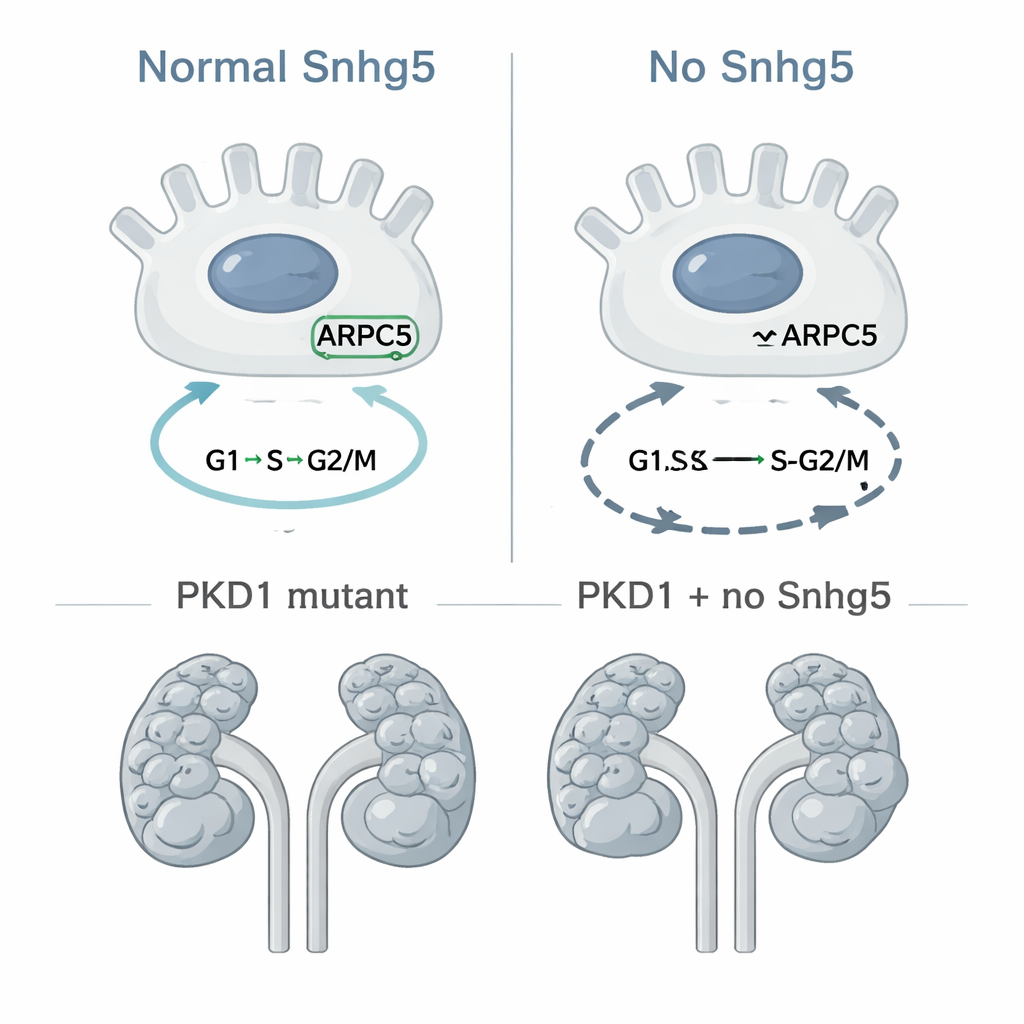
To move beyond correlation, the researchers used CRISPR gene editing to completely delete the Snhg5 gene in mice, creating a global “knockout” line. Contrary to concerns that removing such a strongly altered molecule might be harmful, mice lacking Snhg5 were born at normal ratios, lived normal lifespans, and had kidneys that looked and functioned like those of their healthy littermates. Microscopic examination revealed normal kidney structure without scarring or inflammation, and blood tests showed no signs of impaired kidney filtering. At the molecular level, however, more subtle changes emerged: both mouse kidneys and cultured kidney tubule cells without Snhg5 showed consistent shifts in the activity of genes tied to cell division and DNA copying. In cell culture, more cells stalled in late stages of the cell cycle and in a damaged, sub-G1 state. One protein in particular, ARPC5, part of a complex that helps cells divide by rearranging their internal scaffolding, was reduced when Snhg5 was missing, hinting at a possible chain of cause and effect.
Testing its role in cyst growth directly
Because unchecked cell division is a core feature of PKD, the team next asked if removing Snhg5 would slow cyst formation in a well-established mouse model where the PKD1 gene is disabled specifically in collecting duct cells, the source of many cysts. They bred mice so that some had only the PKD1 mutation, while others lacked both PKD1 and Snhg5. When they examined the animals at 10 days of age, both groups showed severely cystic kidneys, and careful measurements of kidney size, cyst area, and cyst number revealed no meaningful protection from deleting Snhg5. If anything, the double-mutant mice tended to have slightly more cyst burden, although the difference was small and not statistically convincing. In other words, even though Snhg5 influences cell-cycle genes in kidney cells, its absence does not noticeably change how quickly cysts appear or enlarge in this particular PKD model.
What this means for future treatments
For patients and drug developers, the main takeaway is that Snhg5, despite being one of the most strongly altered genetic signals in mouse PKD, is not a linchpin of cyst formation—at least not in the early, fast-growing phase of collecting-duct–derived disease. The gene does seem to fine-tune how kidney cells move through the division cycle, likely through its impact on factors like ARPC5, but this influence is subtle enough that its complete loss leaves kidney development and early PKD progression largely unchanged. These findings underscore a broader lesson: not every striking molecular change in diseased tissue is a promising therapeutic target. Disentangling cause from consequence will require testing long non-coding RNAs like Snhg5 across multiple disease models and time points before they can be confidently pursued as drug candidates.
Citation: D’Amico, S., Dar, U., Eckberg, K. et al. Loss of Snhg5 disrupts cell-cycle regulation without altering cystogenesis in a mouse model of polycystic kidney disease. Sci Rep 16, 4869 (2026). https://doi.org/10.1038/s41598-026-35234-w
Keywords: polycystic kidney disease, long non-coding RNA, Snhg5, cell cycle, kidney cysts