Clear Sky Science · en
Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury
Why heart patients should care about this chemistry
When doctors reopen a blocked heart artery after a heart attack, the rush of fresh blood saves muscle but can also cause extra damage, known as ischemia–reperfusion injury. This study uncovers a surprising culprit inside heart cells: the common metabolic by-product lactate. The authors show that lactate can flip a molecular switch that pushes heart cells toward a specific kind of iron-driven cell death, worsening injury. Understanding this hidden pathway may point to new drugs that better protect the heart during emergency treatment.
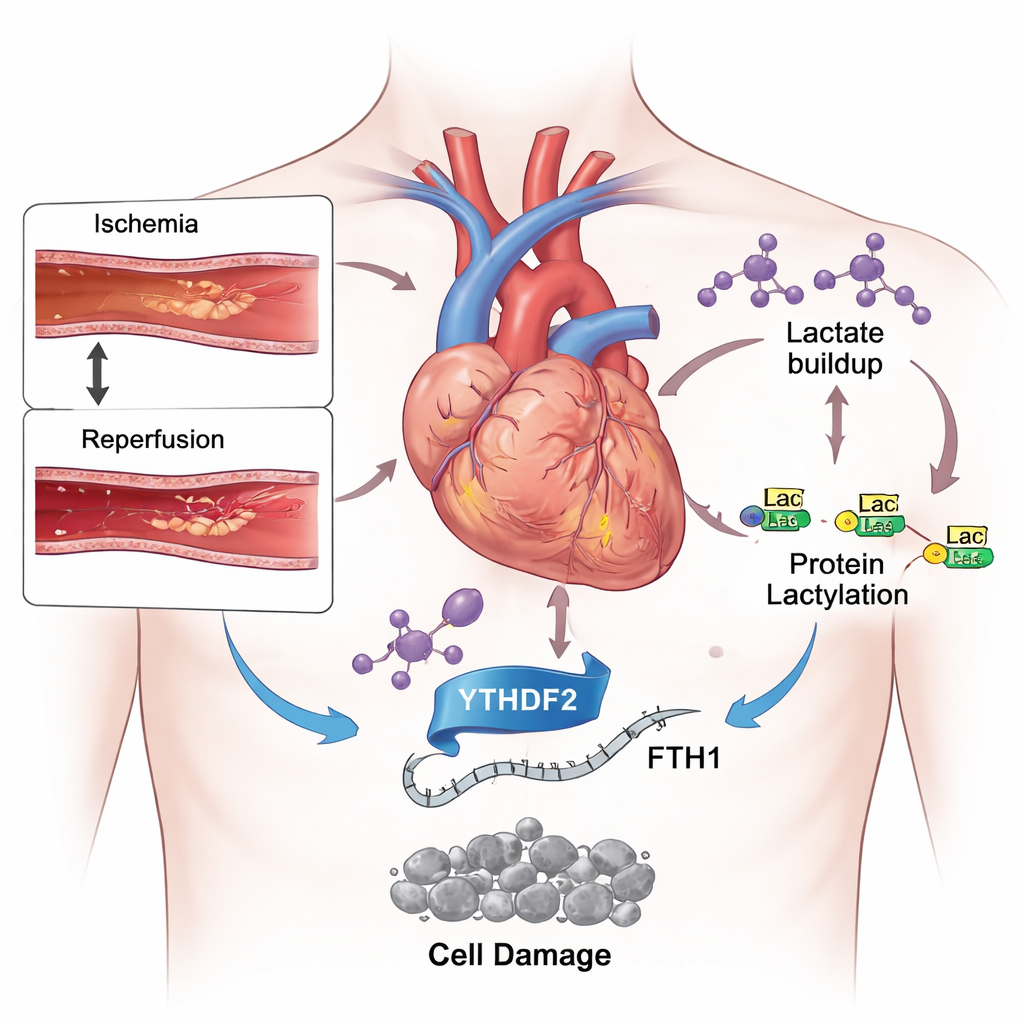
A double-edged sword in heart attack treatment
Modern medicine has become very good at quickly reopening clogged coronary arteries, limiting the initial damage from a heart attack. Yet patients can still lose large areas of heart muscle after blood flow is restored. One reason is that the sudden return of oxygen and nutrients creates a storm of chemical stress inside heart cells. Among several types of cell death triggered in this setting, a newer one called ferroptosis has attracted attention. Unlike more familiar forms such as apoptosis, ferroptosis depends on iron and runaway oxidation of fats in cell membranes, which can permanently weaken the heart.
How lactate becomes more than just “muscle burn”
During a heart attack, starved heart muscle shifts its fuel use toward glycolysis, a backup system that rapidly breaks down sugar but produces large amounts of lactate. Using mice subjected to brief blockage and reopening of a heart artery, and cultured heart-like cells exposed to low oxygen then reoxygenation, the researchers found sharply increased lactate levels. At the same time, they detected more of a chemical tag called lactylation on many proteins and on histones, the scaffolds that organize DNA. When they gave animals a drug that slows glycolysis and reduces lactate production, heart damage shrank, blood markers of injury fell, and the balance between harmful iron and protective antioxidants improved. These results suggest that excess lactate is not just a by-product of stress but an active driver of damage.
A molecular switch that loosens iron’s leash
Diving deeper, the team focused on YTHDF2, a protein that reads chemical marks on RNA and decides how quickly certain messages are destroyed. They discovered that ischemia–reperfusion and added lactate both boosted YTHDF2 levels and increased lactylation around the gene that encodes it, amplifying its production. One of YTHDF2’s key targets turned out to be the RNA for ferritin heavy chain 1 (FTH1), a core part of the cell’s iron storage cage. FTH1 normally tucks iron away in a safe form, preventing it from fueling damaging reactions. In stressed heart cells, YTHDF2 bound more tightly to FTH1 RNA and sped up its decay, leaving cells with fewer ferritin cages, more free iron, heightened oxidative stress, and classic signs of ferroptosis.
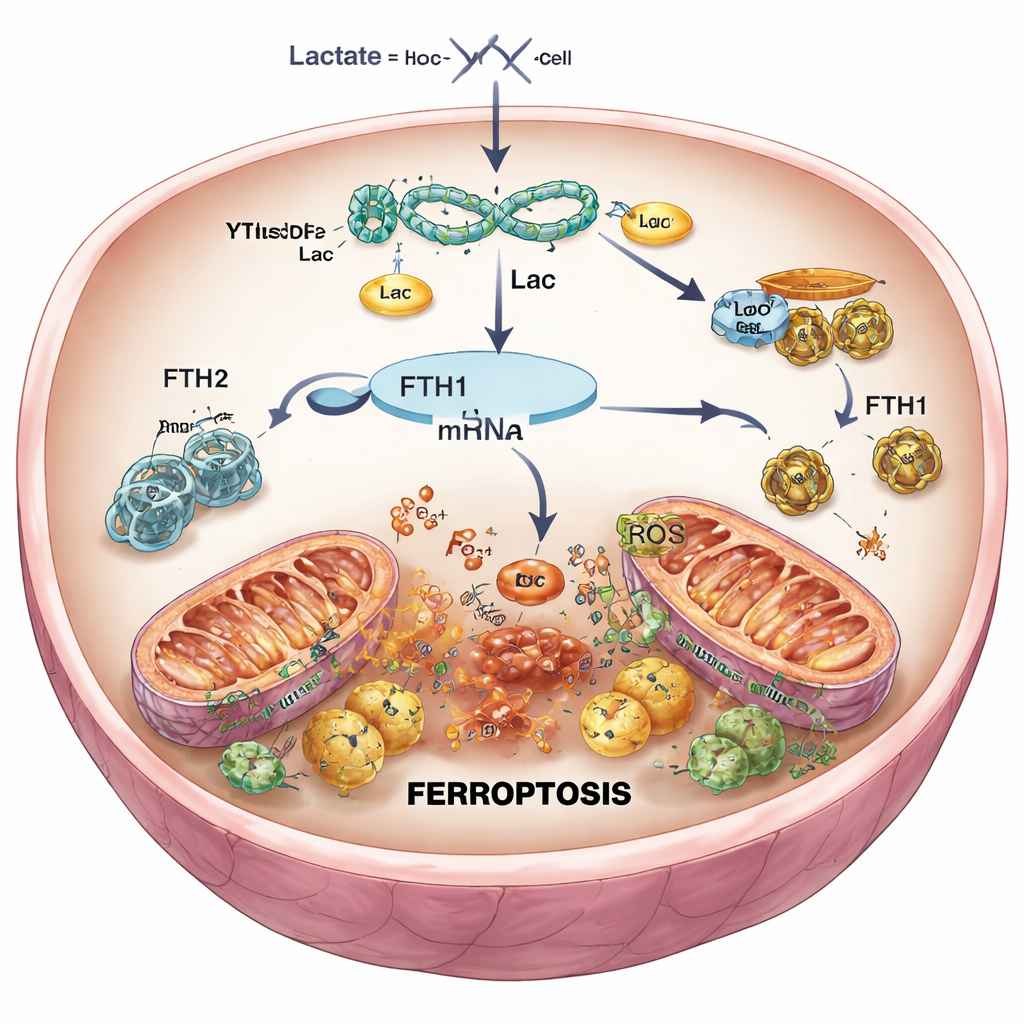
Turning down the death signal in heart cells
To test cause and effect, the researchers used genetic tools to selectively reduce YTHDF2 in heart cells and in mice. When YTHDF2 was knocked down, FTH1 levels rebounded, iron and reactive oxygen species dropped, mitochondria kept a more normal shape, and overall cell survival improved after simulated reperfusion. In mice, less YTHDF2 meant smaller heart attack scars and healthier-looking tissue. However, when FTH1 was simultaneously reduced, these benefits largely disappeared: iron rose again, oxidative damage returned, and infarct size grew. This confirmed that YTHDF2 promotes ferroptosis mainly by suppressing FTH1, loosening control over iron inside heart cells.
What this means for future heart therapies
Putting the pieces together, the study outlines a new chain of events: a blocked and then reopened artery drives lactate buildup; lactate increases YTHDF2 through lactylation; YTHDF2 then destroys the RNA instructions for the iron-guarding protein FTH1; and the resulting iron overload triggers ferroptosis, deepening heart damage. For patients, the message is hopeful: this pathway offers several new points for intervention. Drugs that limit harmful lactate signaling, block the specific modification of YTHDF2, or preserve FTH1 function could make emergency reperfusion safer and protect more heart muscle. While these findings still need confirmation in human tissues, they open a promising avenue toward gentler, more effective treatment for heart attack survivors.
Citation: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Keywords: heart attack, lactate, iron-driven cell death, ischemia reperfusion injury, cardiomyocyte protection