Clear Sky Science · en
A physics-informed graph neural network to approximate docking-based binding affinity for DYRK2 in Alzheimer’s drug repurposing
Why this matters for Alzheimer’s
Alzheimer’s disease is rising worldwide, yet most current drugs only ease symptoms rather than stop the illness. Testing new medicines in the lab is slow and costly, especially for lesser-known brain proteins that might be important in memory and nerve health. This study explores a smart shortcut: using a physics‑aware artificial intelligence model to predict how well existing Alzheimer’s drugs might stick to an underexplored protein called DYRK2, potentially opening new paths for treatment.
A new way to look at old medicines
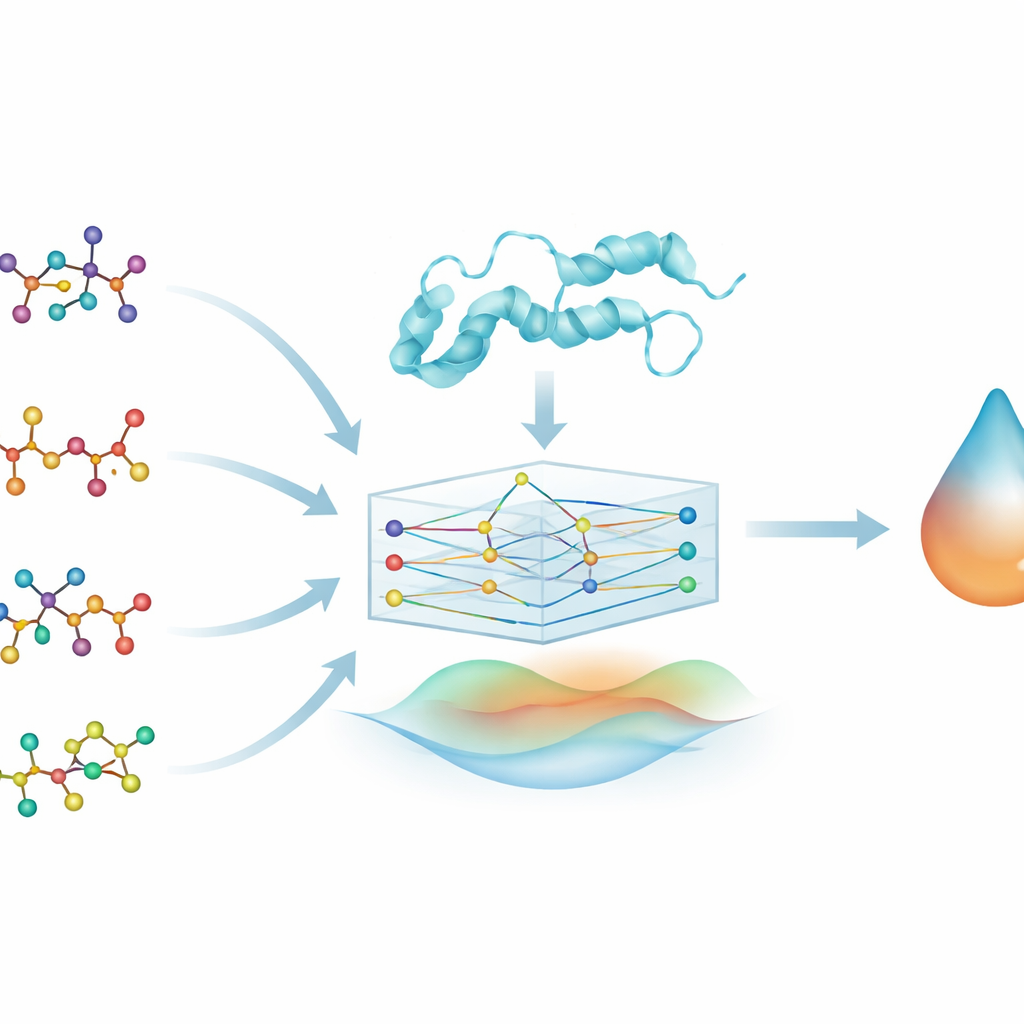
Instead of designing brand‑new compounds from scratch, the researchers focus on drug repurposing—finding fresh uses for medicines that are already approved and known to be relatively safe. They examine four familiar Alzheimer’s drugs (brexpiprazole, donepezil, galantamine, and rivastigmine) and ask how tightly each might bind to DYRK2, a protein kinase involved in nerve cell growth and function. DYRK2 has barely been studied in Alzheimer’s disease, but early evidence links it to synapses, axons, and memory, making it an intriguing target that could complement today’s therapies.
Turning molecules into networks
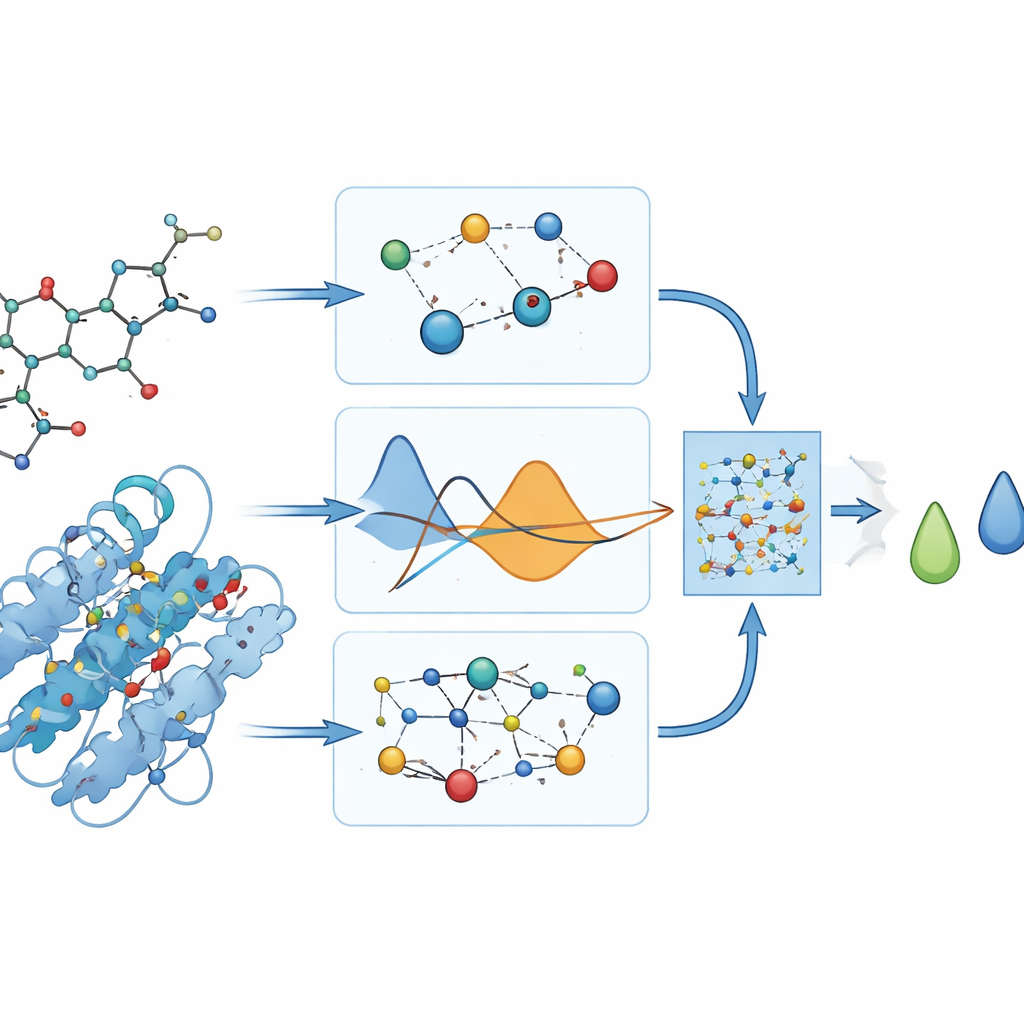
To explore these drug–protein relationships, the team turns each drug molecule into a graph: atoms become points and chemical bonds become lines connecting them. They do something similar for the DYRK2 protein, representing its amino acid sequence as a chain of connected units. A type of machine‑learning model called a graph neural network (GNN) can naturally work with these graph‑shaped inputs, passing information along the connections to learn patterns in shape and chemistry. This allows the model, named PhysDual‑GCN, to “read” both the drug and DYRK2 as interacting networks rather than as simple strings or lists of features.
Blending physics with artificial intelligence
Most deep‑learning tools in drug discovery learn only from data, which can make their inner workings hard to interpret. Here, the authors deliberately weave in basic physical ideas about how atoms interact. Alongside the learned graph features, PhysDual‑GCN calculates two classic energy terms: one capturing electrical attraction and repulsion between partial charges, and another describing the push‑and‑pull of van der Waals forces. These physics‑based energies are combined with the GNN’s internal representation before it outputs a predicted binding strength. In effect, the model is trained to mimic the behavior of standard docking programs—in particular AutoDock Vina and related tools—but to do so faster, while remaining anchored in familiar physical principles.
What the model actually predicts
Because no laboratory measurements exist for how strongly these drugs bind to DYRK2, the authors rely on docking programs to provide “reference” binding scores in units of energy. They carefully avoid feeding those scores into the training process, and instead use them only afterward to judge how well PhysDual‑GCN has learned. For the four Alzheimer’s drugs, the model reproduces the docking values with small average errors (around a third of a kilocalorie per mole) and correctly ranks the compounds: donepezil and brexpiprazole come out as the strongest binders, while galantamine and rivastigmine appear weaker but still reasonably stable. These results show that the physics‑informed GNN can stand in as a computational surrogate for slower docking runs.
Promises and limits of the approach
Despite these encouraging numbers, the authors emphasize that their study has strict boundaries. Only four drugs were examined, and all evaluations rely on other computer programs rather than on real biochemical experiments. The DYRK2 protein is modeled mainly as a one‑dimensional sequence graph, not a full three‑dimensional structure, so the model cannot yet account for the detailed shape of binding pockets. The physical energies themselves are simplified, using standard force‑field parameters and cutoffs. As a result, the work should be seen as a proof‑of‑concept: it shows that physics‑guided graph neural networks can closely track classical docking scores in a low‑data setting, but it does not yet prove that the predictions match reality in the test tube or clinic.
What this means for future Alzheimer’s research
For non‑specialists, the main message is that intelligent, physics‑aware algorithms may help scientists explore new Alzheimer’s targets like DYRK2 far more quickly than traditional methods alone. By highlighting donepezil and brexpiprazole as promising DYRK2 binders and offering a transparent way to approximate docking results, PhysDual‑GCN provides a starting point for deeper laboratory studies. With larger drug libraries, richer 3D protein information, and experimental validation, this kind of model could become a practical tool for screening candidate treatments and guiding drug repurposing efforts aimed at slowing or altering the course of Alzheimer’s disease.
Citation: Gider, V., Budak, C. A physics-informed graph neural network to approximate docking-based binding affinity for DYRK2 in Alzheimer’s drug repurposing. Sci Rep 16, 8357 (2026). https://doi.org/10.1038/s41598-026-35102-7
Keywords: Alzheimer’s disease, drug repurposing, graph neural networks, protein–ligand binding, DYRK2 kinase