Clear Sky Science · en
Glass transition temperatures of pure glass-forming liquids and binary mixtures
Why slowing liquids matter in everyday life
Many materials we rely on—from smartphone screens and plastic packaging to freeze-dried foods and medicines—are technically not true solids but glasses: liquids whose motion has become so slow that they appear frozen. Understanding when a flowing liquid turns into a glass, and how this “freezing” temperature changes when we mix substances, is essential for making products safer, more stable, and longer‑lasting. This paper offers a new way to calculate that key temperature directly from how molecules in a material relax and move, and extends the idea to mixtures such as sugar blends and sugar–water systems used in food and pharmaceuticals.
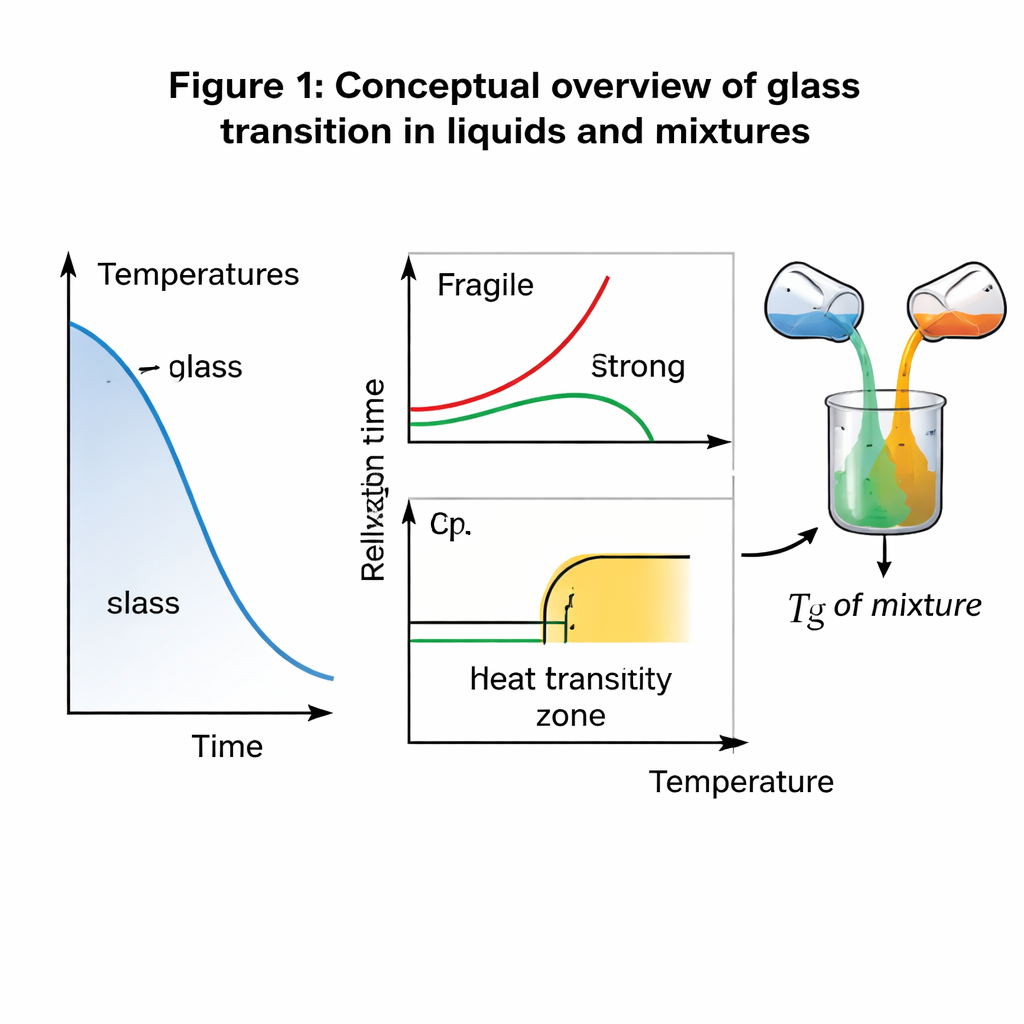
From sharp melts to gradual freezing
Crystals like ice or table salt melt at a clear, sharp temperature. Glasses behave differently. As a glass-forming liquid cools, its molecules gradually slow down until, at the glass transition temperature (Tg), they can no longer rearrange fast enough to keep up with the changing temperature. The material falls out of equilibrium and behaves like a rigid yet disordered solid. Traditionally, scientists have described Tg in two ways: thermodynamically, by a jump in heat capacity (how much heat it takes to warm the material), and dynamically, by the time it takes for molecular rearrangements. A common rule of thumb says Tg is where the structural relaxation time reaches around 100–1000 seconds—but this is mainly a convenient convention rather than a principle grounded in physics.
A cleaner link between time, temperature, and scanning rate
The authors build on a classic idea that directly connects the rate at which temperature is changed in an experiment (the scan rate) to how quickly the material’s relaxation time is changing with temperature. In essence, the glass transition is defined as the point where the timescale of structural relaxation becomes comparable to the timescale of the temperature scan. Using standard models that describe how relaxation time depends on temperature, they turn this condition into explicit mathematical equations for Tg. These equations involve a special mathematical function (the Lambert W function) that has recently become widely available in scientific software, making it practical to solve such problems analytically rather than purely by numerical fitting.
Why the “universal” glass timescale is a myth
With their new equations, the authors show that the relaxation time at the glass transition, often assumed to be a fixed “laboratory” value, in fact depends strongly on both the scan rate and the material’s activation energy—the effective energy barrier that controls molecular motion. For a given scanning speed, materials with higher activation energies or higher Tg values can have glass-transition relaxation times that differ by orders of magnitude. Simulations using widely used glass-transition models confirm that, although different ways of defining Tg (such as where the heat‑capacity curve bends most sharply) are not identical, they give very similar temperatures, while clearly demonstrating that there is no single, universal relaxation time that applies to all glass formers.
How mixtures of glass formers share their traits
Real‑world materials are rarely pure. In polymer blends, food products, or amorphous drugs, two or more glass-forming substances are mixed, and manufacturers need to know how the mixture’s Tg depends on composition. Empirically, this is often described by the Gordon–Taylor equation, which uses a fitting constant whose physical meaning has been unclear and debated. The authors propose a dynamic alternative: they assume that key kinetic parameters—such as effective activation energies and related quantities—mix in a simple way based on the mass fractions of each component. From these “ideal dynamic mixing rules,” they derive a general expression for the mixture Tg and show that, in a limiting case, the familiar Gordon–Taylor formula naturally appears, with the fitting constant linked to the components’ activation energies or fragilities (a measure of how sharply their relaxation slows on cooling).
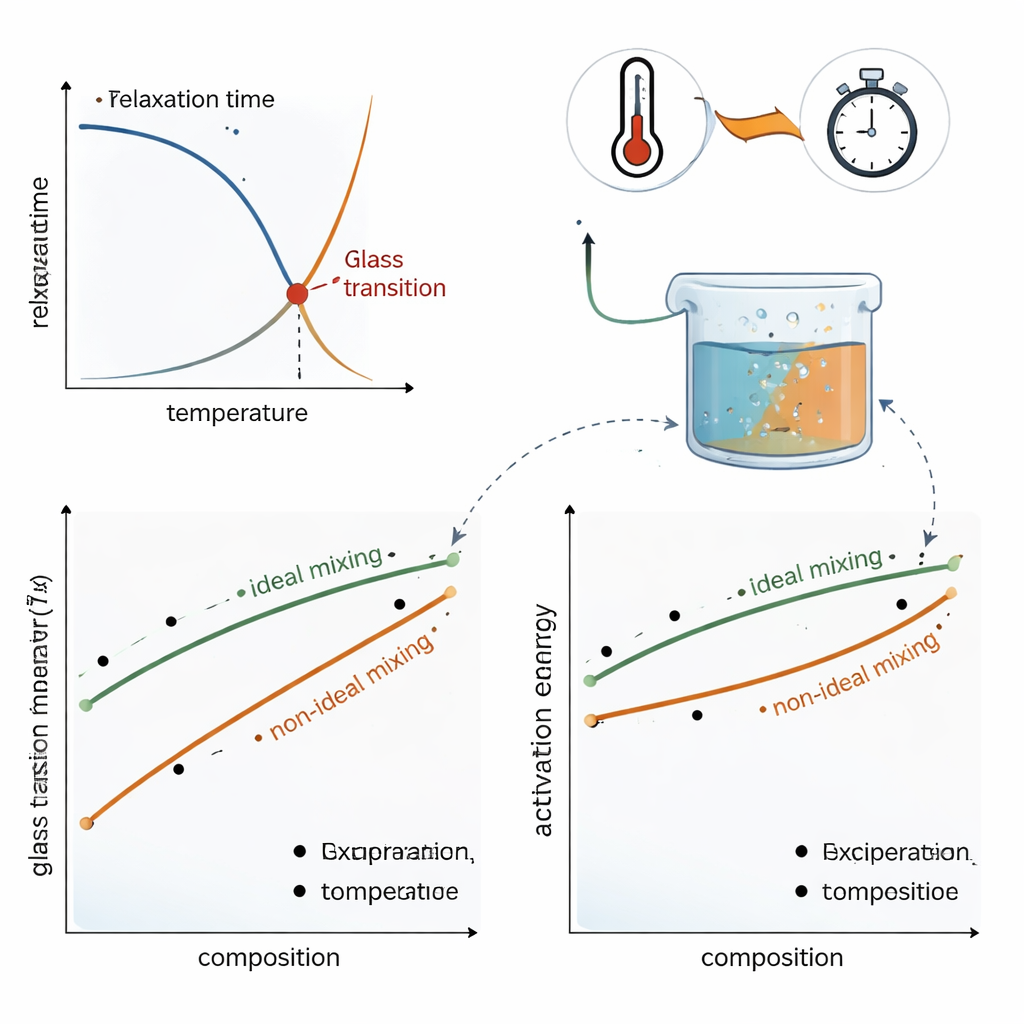
Real mixtures: when ideal rules break down
To test their framework, the authors examine data from two practically important systems. In mixtures of the sugars sucrose and trehalose—common in food and biological preservation—the measured Tg and activation energy vary only slightly from what ideal dynamic mixing would predict; modest adjustments to the mixing rules capture the observed curves. In sucrose–water mixtures, however, the behavior is strongly non‑ideal: adding even small amounts of water decreases the activation energy and Tg much more than a simple average would suggest. By allowing the mixing rules to be nonlinear, the new model can reproduce the full, curved dependence of Tg and activation energy on composition, reflecting how water dramatically loosens the molecular network of the sugar glass.
Take‑home message for materials and everyday products
Put simply, this work shows that the temperature at which a liquid becomes a glass is not governed by a single magic timescale, but by how quickly the liquid’s internal motions respond to a given cooling or heating rate. The same kinetic logic extends naturally to mixtures, where the widely used Gordon–Taylor relation emerges as a special case of more general dynamic rules. For technologists designing tougher phone screens, longer‑lasting foods, or more stable medicines, this framework offers a more physically grounded way to predict and tune glass transition temperatures across both pure materials and complex blends.
Citation: Kocherbitov, V., Argatov, I. Glass transition temperatures of pure glass-forming liquids and binary mixtures. Sci Rep 16, 1317 (2026). https://doi.org/10.1038/s41598-026-35024-4
Keywords: glass transition, relaxation time, fragility, glass-forming mixtures, Gordon–Taylor equation