Clear Sky Science · en
Phenomenological model of transthyretin stabilization
Why this matters for patients and families
Transthyretin amyloidosis is a serious condition in which a normal blood protein, transthyretin (TTR), falls apart and its pieces can clump into harmful deposits in the heart and nerves. New drugs such as tafamidis and acoramidis are designed to keep this protein in its safe, four-part form, and they have already improved outcomes for many people. Yet doctors see a puzzling effect in treated patients: blood levels of TTR rise by more than 30%, and it is not obvious why. This paper uses a stripped-down, mathematics-based model to explore what might be happening inside the body and what that means for how these medicines really work. 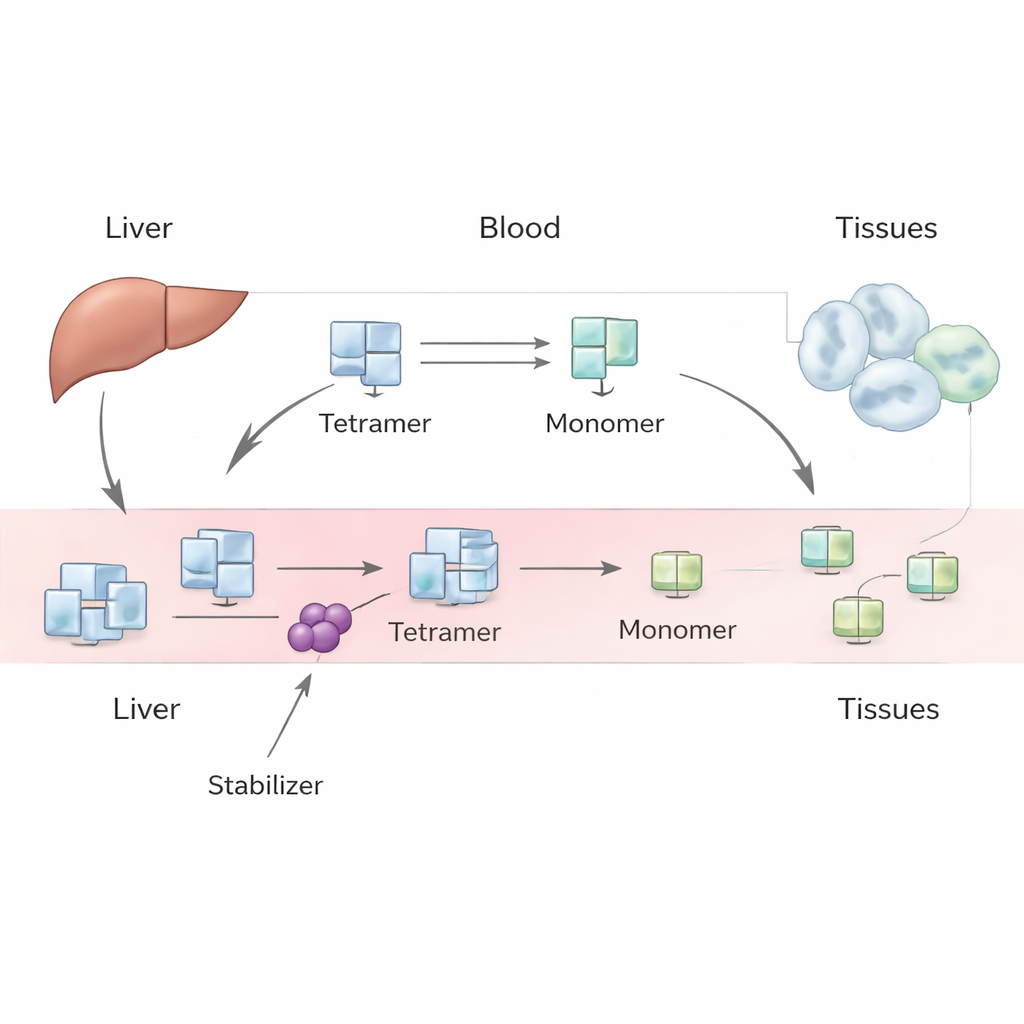
A protein that can help or harm
TTR is made mainly in the liver and normally circulates in the blood as a stable bundle of four identical subunits, known as a tetramer. It transports thyroid hormone and vitamin A. Under some conditions, including aging or inherited gene variants, this tetramer can come apart into single subunits, or monomers. These monomers can misfold and stick together as amyloid fibrils, which damage tissues and lead to transthyretin amyloidosis of the heart (cardiomyopathy) or nerves (neuropathy. Drugs like tafamidis and acoramidis are designed to bind the tetramer and make it harder for it to fall apart, slowing disease progression. However, when patients start these drugs, their measured TTR levels in blood reliably rise, and this increase is larger than simple lab experiments would suggest.
Building a simple picture of a complex system
The authors tackle this puzzle with a phenomenological model—one that focuses on overall observable behavior rather than every microscopic detail. In their framework, the liver produces TTR tetramers at a steady rate, which then enter the bloodstream. Once in circulation, tetramers can dissociate into monomers and reassemble, and both tetramers and monomers can be removed from blood by uptake into tissues and degradation. By writing down a pair of mass-balance equations for tetramers and monomers, the team explores different scenarios: one in which monomers mostly rejoin tetramers, one in which they are quickly removed, and an intermediate case where both processes matter. They use historical human tracer studies and modern lab data to estimate key quantities, such as how fast tetramers are cleared, how quickly they fall apart, and how strongly drugs slow this breakup.
Why tetramer stabilization alone is not enough
Armed with these estimates, the researchers ask a pointed question: if a drug could perfectly prevent tetramers from breaking apart, how much would the blood TTR level rise? Across all plausible regimes, the answer is modest—on the order of 15% at most for typical parameter values, and often less, depending on how monomers are handled. This falls far short of the >30% increase observed in treated patients. The mismatch remains even if one allows for generous uncertainty in the known parameters. The model therefore suggests that simply slowing tetramer breakup cannot, by itself, explain the full clinical effect. Other processes that control how quickly TTR is made, taken up by cells, or broken down must also be changing when stabilizer drugs are present. 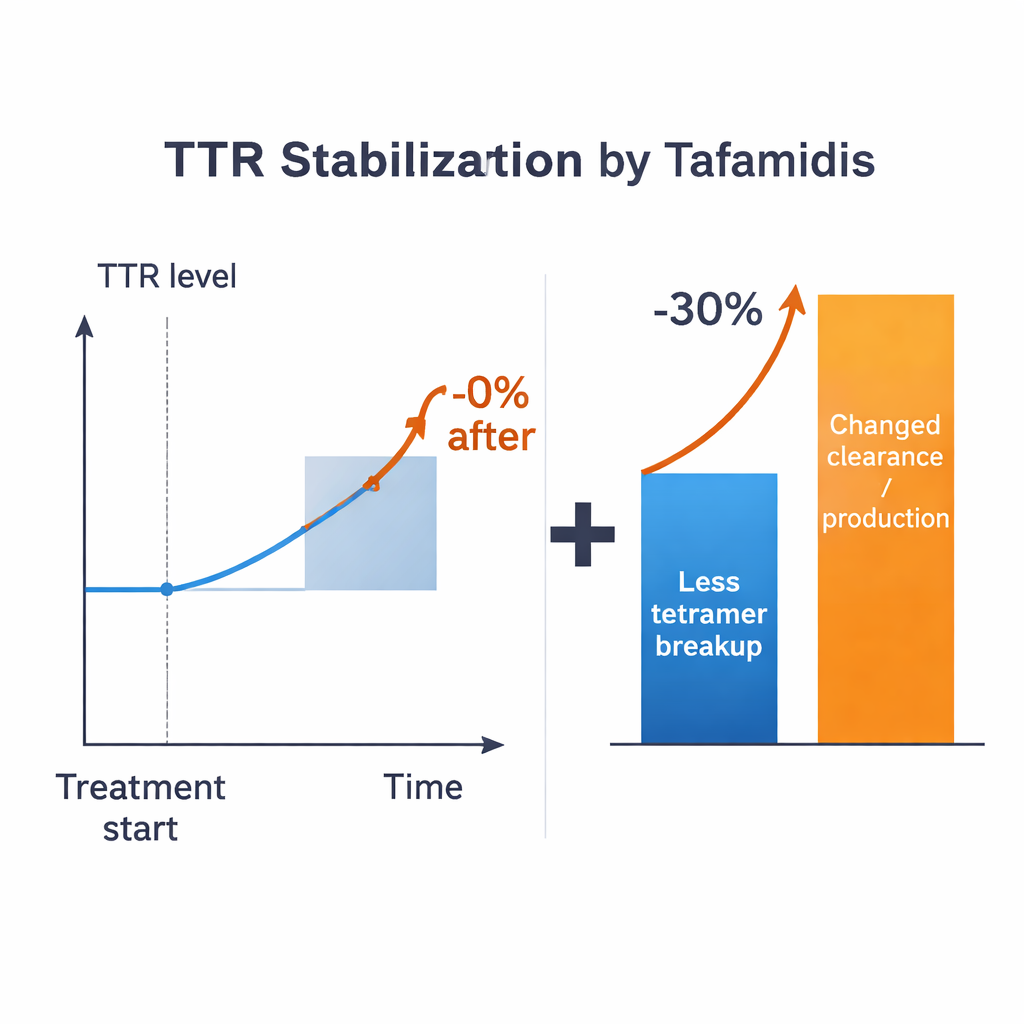
Clues from drug levels and clever experiments
To connect drug exposure to TTR behavior, the authors combine a basic pharmacokinetic model of tafamidis (how the drug moves through the body over time) with specialized “subunit exchange” assays. In these experiments, labeled and unlabeled TTR tetramers are mixed in human plasma, and the gradual swapping of subunits reveals how often tetramers fall apart. Measuring this process at different drug concentrations yields a direct, data-driven link between tafamidis level and effective tetramer stability, without needing to know how much drug is bound to albumin or thyroid hormone. This phenomelogical link feeds into the model and cleanly sidesteps many biochemical unknowns. Yet, even with this advantage, the calculations still cap the expected rise in TTR at roughly half the clinically observed increase, reinforcing the idea that changes in clearance, internalization, degradation, or even synthesis must be part of the story.
What this means going forward
For non-specialists, the key message is that these stabilizer drugs are likely doing more than just “gluing” TTR tetramers together. They probably also affect how the body produces, removes, or recycles the protein. The authors argue that simple, transparent models like theirs are powerful because they expose such gaps in our understanding and point to concrete experiments—for example, directly measuring how fast monomers are cleared, how quickly different TTR forms are taken up by cells, or whether drug-bound TTR is handled differently than unbound protein. Better answers to these questions would not only refine treatment of transthyretin amyloidosis but might also reveal general rules about other diseases in which normal proteins turn into harmful aggregates.
Citation: Lisowski, B., Ulaszek, S., Wiśniowska, B. et al. Phenomenological model of transthyretin stabilization. Sci Rep 16, 4904 (2026). https://doi.org/10.1038/s41598-026-35000-y
Keywords: transthyretin amyloidosis, protein stabilization, tafamidis, pharmacokinetic modeling, amyloid diseases