Clear Sky Science · en
Graph-theoretic analyses of saturation fraction of repulsive dopants in solid solutions
Why packed-in atoms matter
Modern metals and semiconductors are rarely pure. Engineers deliberately mix in different kinds of atoms—called dopants—to tune strength, toughness, corrosion resistance, or electronic behavior. But in many important materials, these dopant atoms actively avoid one another, preferring not to sit next to the same kind of atom. This quiet game of atomic “social distancing” turns out to limit how much of a dopant a material can safely and usefully hold. The paper explores this limit using tools from mathematics and physics, and shows that surprisingly simple rules about the underlying atomic grid can predict when repulsive dopants reach their saturation point.
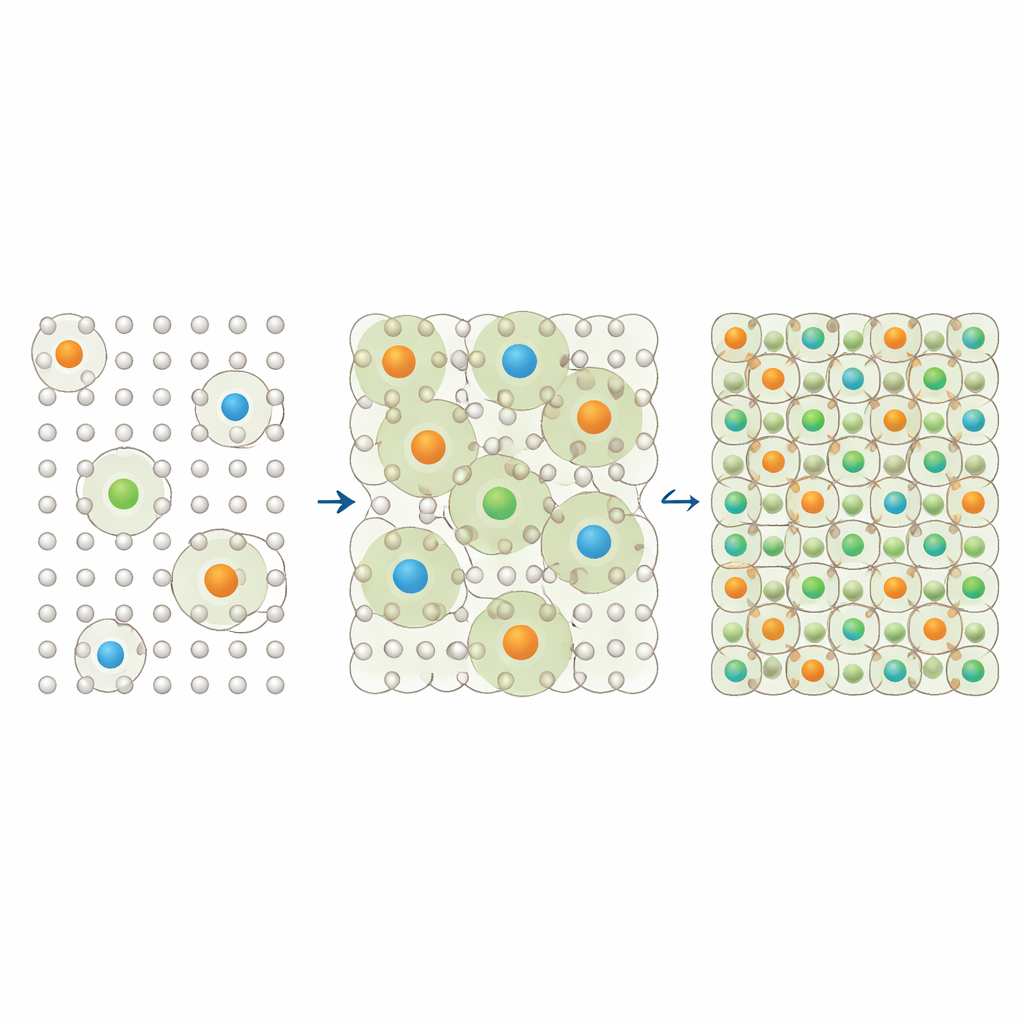
Atoms on a grid
The authors focus on substitutional solid solutions, a broad class of alloys where each point in a regular atomic grid (a lattice) is occupied either by a base atom or by a dopant atom. Experiments have shown that in many systems—such as iron–chromium steels, complex high‑entropy alloys, and group‑IV semiconductor alloys like germanium–tin—certain dopant pairs hardly ever sit next to each other. Instead, they form patterns known as short‑range order, where local arrangements are biased away from random. This hidden ordering can strongly affect mechanical and electrical properties, yet is hard to see directly in experiments. A natural, but previously unanswered, question is: if dopant atoms must avoid being neighbors, how many can we fit into the lattice before that rule becomes impossible to satisfy?
A simple packing game on a lattice
To address this, the researchers model dopant insertion as a random packing process on a lattice. They imagine starting with a pure base material and adding dopant atoms one by one. Each new dopant is placed randomly on a site that is not already a dopant and not a neighbor of any dopant. Once selected, a site becomes a dopant site; its neighboring sites become blocked for future dopants. This process continues until there are no remaining eligible sites. The final fraction of sites occupied by dopants is defined as the saturation fraction. Using computer simulations on 14 different lattice types—including common structures like body‑centered cubic (found in steels), face‑centered cubic, and more exotic high‑dimensional lattices—the authors show that each lattice has a very reproducible saturation fraction, an intrinsic fingerprint of how it accommodates repulsive dopants.
Graphs, connections, and a universal rule
Instead of treating each lattice separately, the authors recast the problem using graph theory, where each atomic site is a point (vertex) and each neighbor relationship is a link (edge). They approximate real lattices by random regular graphs—networks in which every point has the same number of neighbors, called the coordination number. They then write simple equations that track, step by step, how many sites are dopants, blocked neighbors, or still available as dopants during the packing process. Solving these equations yields a compact formula that predicts the saturation fraction solely from the coordination number. Simulations on large random graphs confirm this prediction without any adjustable parameters, showing that the saturation of repulsive dopants is, to first order, controlled just by how many neighbors each site has.
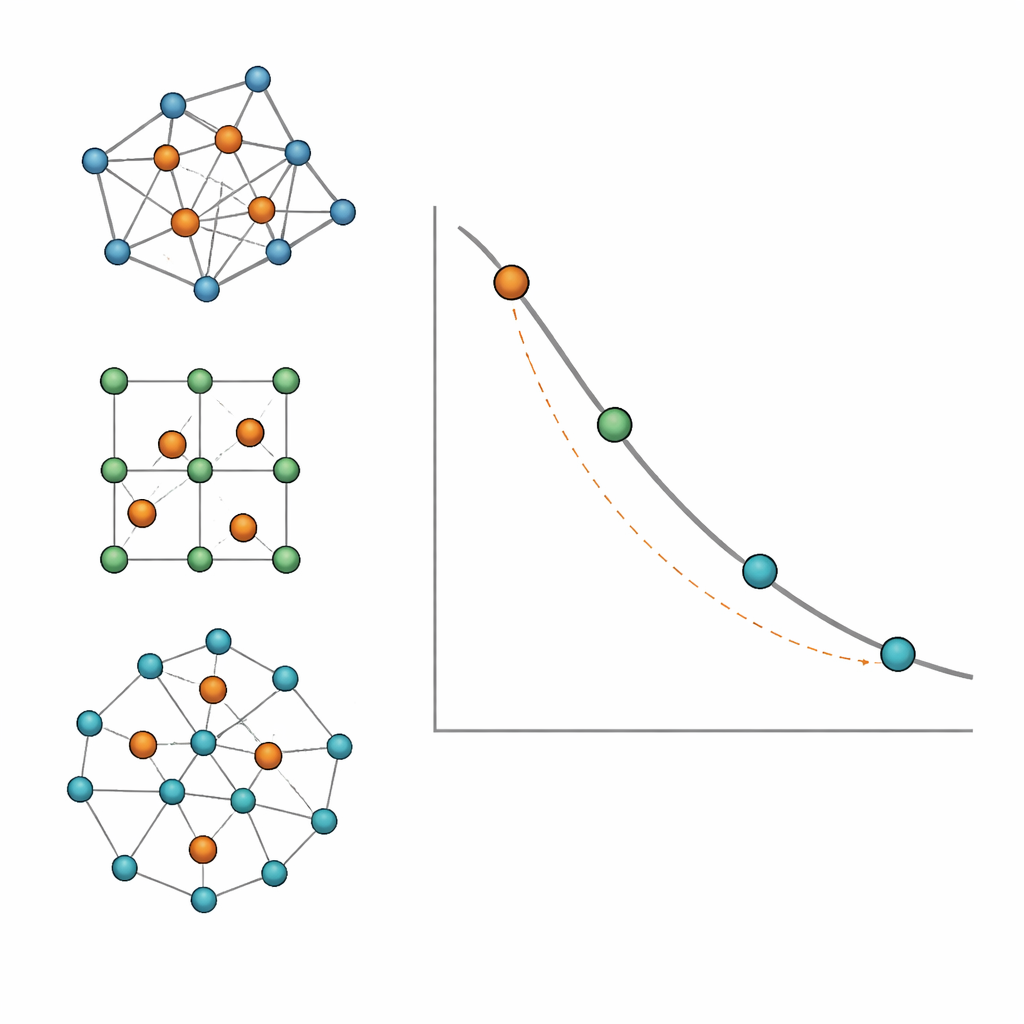
When local loops change the limit
Real crystals, however, are not perfectly random networks. They contain many small loops of connected sites—triangles, squares, hexagons—which subtly alter the packing capacity. To capture this, the authors turn to another graph property called girth: the size of the smallest loop in the network. By comparing simulations on real lattices with the random‑graph formula, they find a systematic pattern. Lattices rich in three‑site loops (girth 3), like the face‑centered cubic structure, tend to have lower saturation fractions than predicted. Lattices dominated by four‑site loops (girth 4), such as simple cubic and body‑centered cubic, can pack repulsive dopants more densely than the random‑graph model suggests. Structures with larger loops sit closer to the simple prediction. Even one‑dimensional chains and finite rings fit neatly into this graph‑theoretic picture.
From abstract graphs to real materials
These insights have concrete consequences. In ferritic stainless steels, chromium atoms repel one another when dilute; if their concentration exceeds the saturation fraction for the body‑centered cubic lattice, chromium‑rich clusters that embrittle the steel are more likely to form. In high‑entropy and medium‑entropy alloys, the number of elements and their fractions determine whether repulsive species can remain non‑neighboring at all; for a body‑centered cubic alloy, for example, a four‑element mixture can stay below the saturation threshold, while a three‑element one cannot. The same ideas extend to hydrogen occupying interstitial sites in metals and even to disordered systems like metallic glasses, as long as approximate connectivity and loop sizes are known.
What this means in plain terms
In essence, the study shows that there is a mathematically predictable ceiling on how many mutually avoiding dopant atoms a material can hold, and that this ceiling depends mostly on how many neighbors each site has and how those neighbors form small loops. By blending detailed simulations with a simple graph‑based model, the authors provide a universal recipe for estimating this saturation fraction across many different materials. For engineers, this means that safe and effective dopant levels—before unwanted clustering or electronic changes emerge—can be estimated from a small set of structural features, offering a powerful new handle for designing advanced alloys and semiconductors.
Citation: Kubo, A., Abe, Y. Graph-theoretic analyses of saturation fraction of repulsive dopants in solid solutions. Sci Rep 16, 7650 (2026). https://doi.org/10.1038/s41598-025-30829-1
Keywords: repulsive dopants, short-range order, random graphs, alloy design, saturation fraction