Clear Sky Science · en
Annotation of 200 Insect Genomes with BRAKER for Consistent Comparisons across Species
Why Insect Genomes Matter
Insects shape our world: they pollinate crops, spread diseases, recycle nutrients, and inspire new materials and technologies. Today we can read the DNA of thousands of insect species, but simply having their genomes is not enough. We also need a clear map of where each gene lies and what it likely does. This article describes a large, standardized effort to annotate the genes of 200 insect species using an automated workflow called VARUS-BRAKER, making it much easier for scientists to compare species and uncover how insects evolved their remarkable diversity.
The Problem of Unfinished Genetic Maps
Over the last two decades, insect genome sequencing has exploded from about twenty species to more than four thousand. Yet only about one in ten of these genomes has a proper gene annotation in public databases. Even when annotations exist, many were created years ago with older methods and limited data. Different research groups often used different software and evidence, which can create artificial differences: a gene may appear to be missing or oddly shaped in one species simply because it was annotated with another tool. This patchwork of methods makes it risky to draw conclusions about how insect genes truly differ across species.
A One-Button Workflow for Many Species
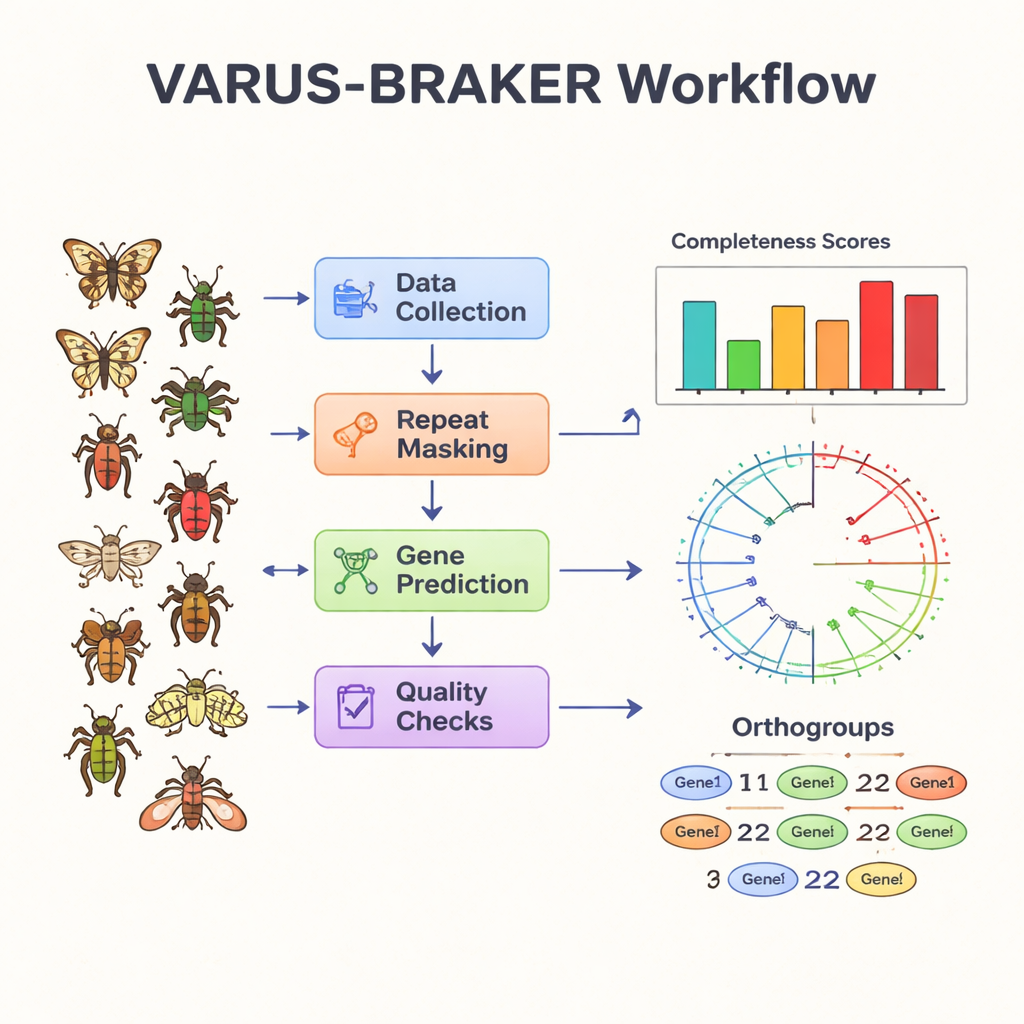
The authors address this bottleneck by building an automated workflow centered on the BRAKER3 gene-prediction pipeline. Their VARUS-BRAKER system is designed so that, in the easiest mode, a user needs to provide only the scientific name of a species. The workflow then automatically downloads the best available genome from public archives, collects matching RNA sequencing data that show which genes are active, and retrieves protein information from related species. It masks repetitive DNA, aligns RNA reads to the genome, and combines RNA and protein “clues” to teach its models where genes are likely to start, stop, and splice. Quality checks like BUSCO and OMArk then assess how complete and clean the resulting gene set is.
A Broad Tour Across the Insect Tree
Using this system, the team annotated 200 insect genomes chosen to cover the main branches of the insect family tree, with a focus on holometabolous insects—those with complete metamorphosis from larva to pupa to adult—plus a diverse set of relatives. Their sample spans 77 families and 14 orders, including flies, butterflies, beetles, bees, ants, aphids, cockroaches, and others. Eighty-five of these species had no prior annotation in GenBank. For each species, the workflow predicted protein-coding genes, resulting in more than 4.2 million protein sequences. Most genomes and their predicted proteomes passed stringent completeness tests, typically reaching at least 85–95% coverage of expected core genes, indicating that the automated approach produces high-quality results.
From Gene Lists to Biological Meaning
Listing genes is only part of the story; researchers also need hints about what these genes do. To that end, the authors applied a functional annotation pipeline called FANTASIA, which uses modern protein language models to assign Gene Ontology (GO) terms—standard labels for biological roles—to each protein. Compared with the widely used InterProScan tool, FANTASIA annotated about 1.6 times more proteins, while still agreeing closely when both methods made predictions. The team also grouped related genes into “orthogroups,” sets of genes that share a common ancestor, and used these to build an evolutionary tree of the 200 species. This framework makes it possible to ask which genes are shared, lost, or expanded in different insect lineages, and to connect gene repertoires to traits such as metamorphosis or larval behavior.
A Reusable Resource for Future Discoveries
All data from this project—including gene structures, protein sequences, functional labels, orthogroups, species trees, and tRNA predictions—are freely available through public repositories. The authors also publish the full VARUS-BRAKER workflow as open-source code so other scientists can annotate new insect genomes, or even other animals and plants, in a consistent way. For non-specialists, the key takeaway is that this work turns a scattered collection of DNA sequences into a coherent, comparable atlas of insect genes. With these standardized maps, future studies can more reliably uncover how insects evolved flight, metamorphosis, and ecological success, and can better target genes relevant to agriculture, conservation, and disease control.
Citation: Saenko, S., Hoff, K.J. & Stanke, M. Annotation of 200 Insect Genomes with BRAKER for Consistent Comparisons across Species. Sci Data 13, 288 (2026). https://doi.org/10.1038/s41597-026-06840-0
Keywords: insect genomics, genome annotation, comparative genomics, evolutionary biology, bioinformatics