Clear Sky Science · en
Minimum virtual dataset for reproducible triploid de novo genome assembly
Why three-copy genomes matter
Many crops and other organisms don’t carry just two copies of each chromosome, like humans do—they can carry three or more. Piecing together these extra copies from DNA sequencing data is surprisingly difficult, because the copies are very similar yet not quite identical. This article introduces a small but carefully designed “virtual” dataset that lets researchers test and compare genome-assembly software on a realistic three-copy (triploid) problem, under conditions that are completely known and reproducible.
Building a simple stand‑in genome
Instead of starting from a real plant or animal, the author first creates a random stretch of DNA one million letters long to act as a clean template. This template is then duplicated into three separate versions, standing in for the three chromosome sets in a triploid organism. To mimic how real genomes slowly change over time, the study introduces a fixed number of tiny changes—single-letter substitutions—step by step into each copy. Repeating this process over 100 steps produces triplets of genomes that range from almost identical to clearly but still moderately different. This controlled “divergence gradient” is the backbone of the benchmark.
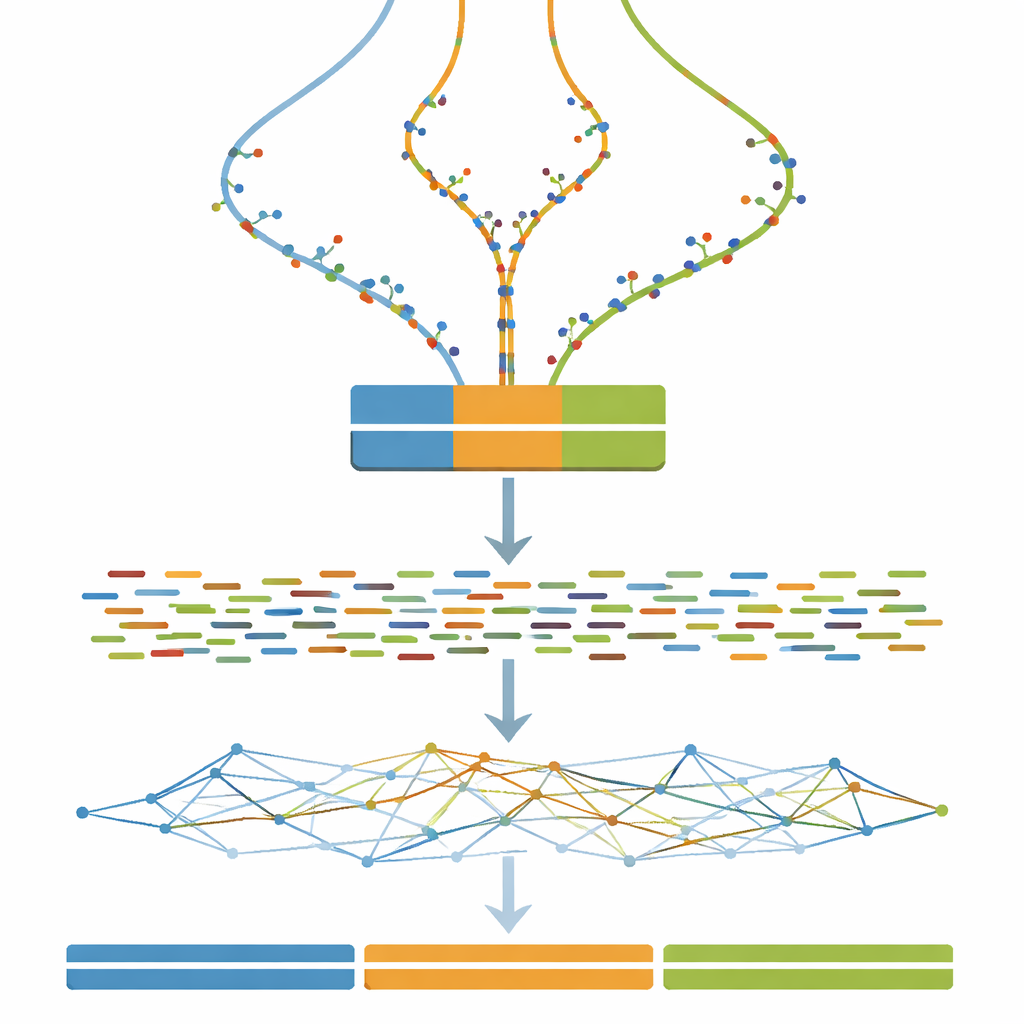
Turning virtual genomes into virtual experiments
Once each three-copy genome is defined, the next step is to imitate what a DNA sequencing machine would see. The study uses widely adopted software to simulate short paired DNA fragments, similar to those produced by an Illumina sequencer, at a constant and fairly high depth of coverage. Optional cleanup steps mimic common real-world practices such as correcting random sequencing errors and merging overlapping read pairs. As a result, anyone using the dataset can test not only their assembly algorithms, but also how typical pre-processing choices influence the final assembled genomes.
Stress-testing assembly strategies
The heart of the work is a giant experiment in which all simulated reads are fed into a single genome-assembly program while changing only one key setting: the k-mer size, a parameter that controls how finely the software “chunks” the reads when reconstructing the genome. For every combination of divergence level (from 0 to 100 steps) and k-mer size (a wide range of odd values), a fresh assembly is built. A companion evaluation tool then measures how continuous the assembled pieces are, how many pieces exist, and how closely their combined length matches the known three-million-letter truth. These measurements are summarized as heatmaps, revealing broad zones where assemblies collapse different copies into one, fragment into many small pieces, or come close to the ideal of three long, accurate contigs.
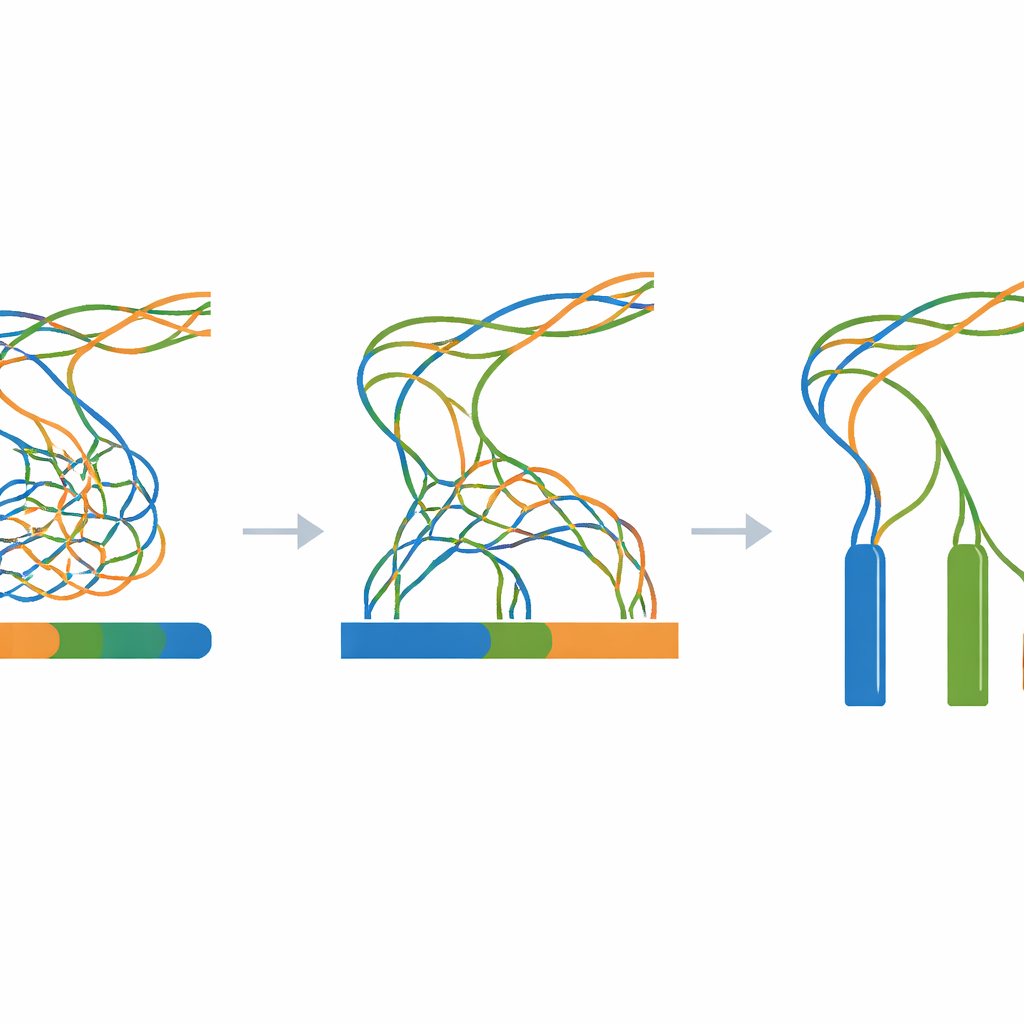
A transparent reference for tricky genomes
Because every stage is synthetic and scripted—from the initial random template to the final assemblies—researchers can reproduce the entire workflow on any standard Linux computer using only open-source tools. The Zenodo archive linked in the paper contains the template genome, all intermediate mutated sequences, all simulated reads, and every assembly result, along with logs and simple helper scripts. Technical checks confirm that the mutation process behaves as expected, that the simulated reads match the requested lengths and coverage, and that assemblies show the anticipated pattern: strong over-collapsing when the three copies are nearly identical, and clearer separation as they drift further apart.
What this means in plain terms
In everyday language, this article offers a controlled test track for software that tries to rebuild three similar instruction books from piles of shuffled fragments. By gradually increasing how different the three books are, and by systematically changing a key setting in the reconstruction process, the dataset makes it easy to see when and how current methods fail or succeed. Developers can use it to tune new algorithms, while users can better understand which settings work best for triploid genomes. Although the DNA itself is artificial, the lessons it enables—about collapse, separation, and the impact of parameter choices—are directly relevant to real-world efforts to decode the complex genomes of many important species.
Citation: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Keywords: triploid genome assembly, polyploid benchmarking, synthetic DNA dataset, de novo assembly, k-mer optimization