Clear Sky Science · en
Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement
Why your medical data shouldn’t end up in a digital graveyard
Modern medicine increasingly relies on huge datasets that describe the thousands of proteins at work in our cells. These files are often shared openly online, promising that other scientists can double‑check results or ask new questions without running fresh experiments. But if the data are posted in confusing formats, missing key details, or tied to proprietary software, they become “data tombs”: visible to everyone, yet practically unusable. This article shows how a university course turned students into data detectives to expose this hidden problem—and suggests simple fixes that could make shared data truly reusable.
Learning science by redoing real studies
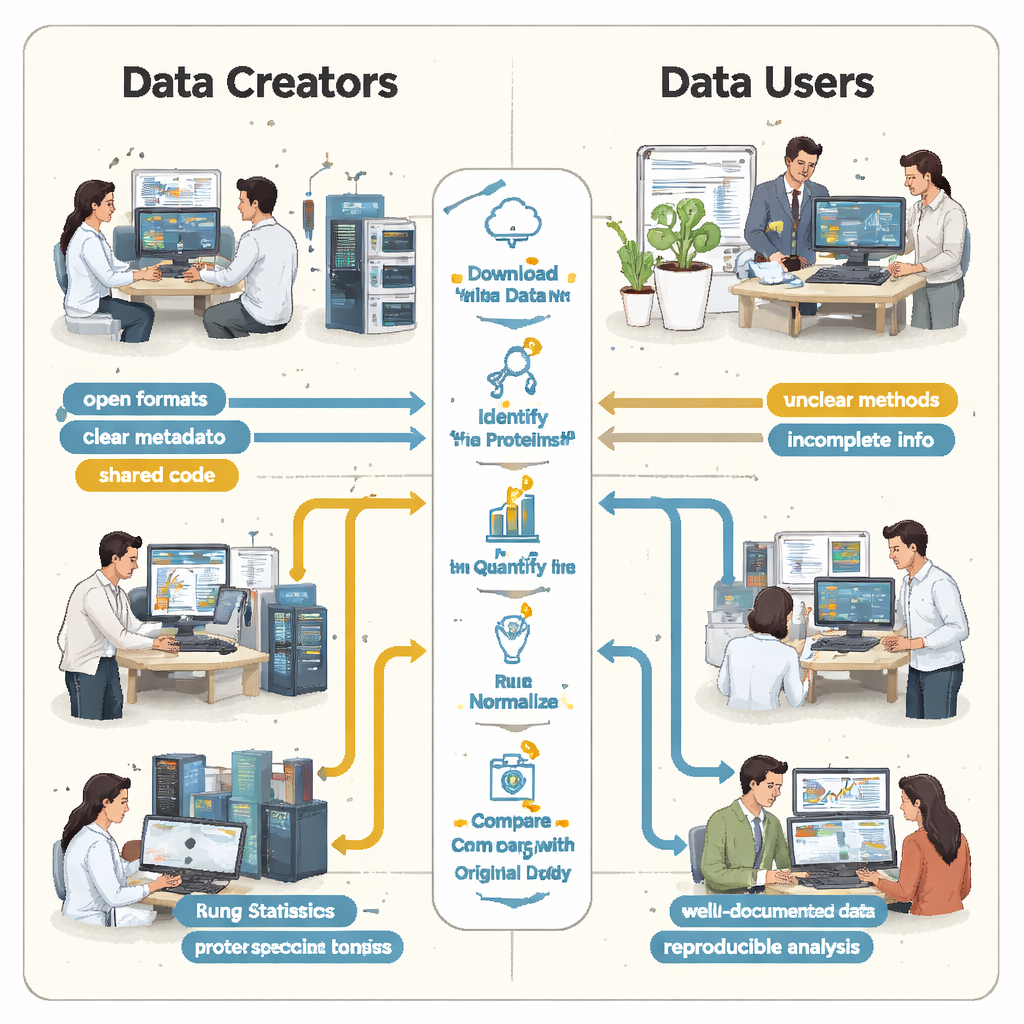
At the University of Helsinki, graduate students in a mass‑spectrometry proteomics course were asked to do something ambitious: pick real, publicly available protein datasets from a major repository and try to reproduce the published findings. Working in small teams, they downloaded six projects from the ProteomeXchange network, which hosts mass‑spectrometry results from many labs worldwide. Using a shared analysis pipeline in the R programming language, the students followed the same broad steps as the original researchers: identify proteins, measure their abundance, clean up the data, and test which proteins change between conditions such as disease versus healthy tissue.
Big promises, missing instructions
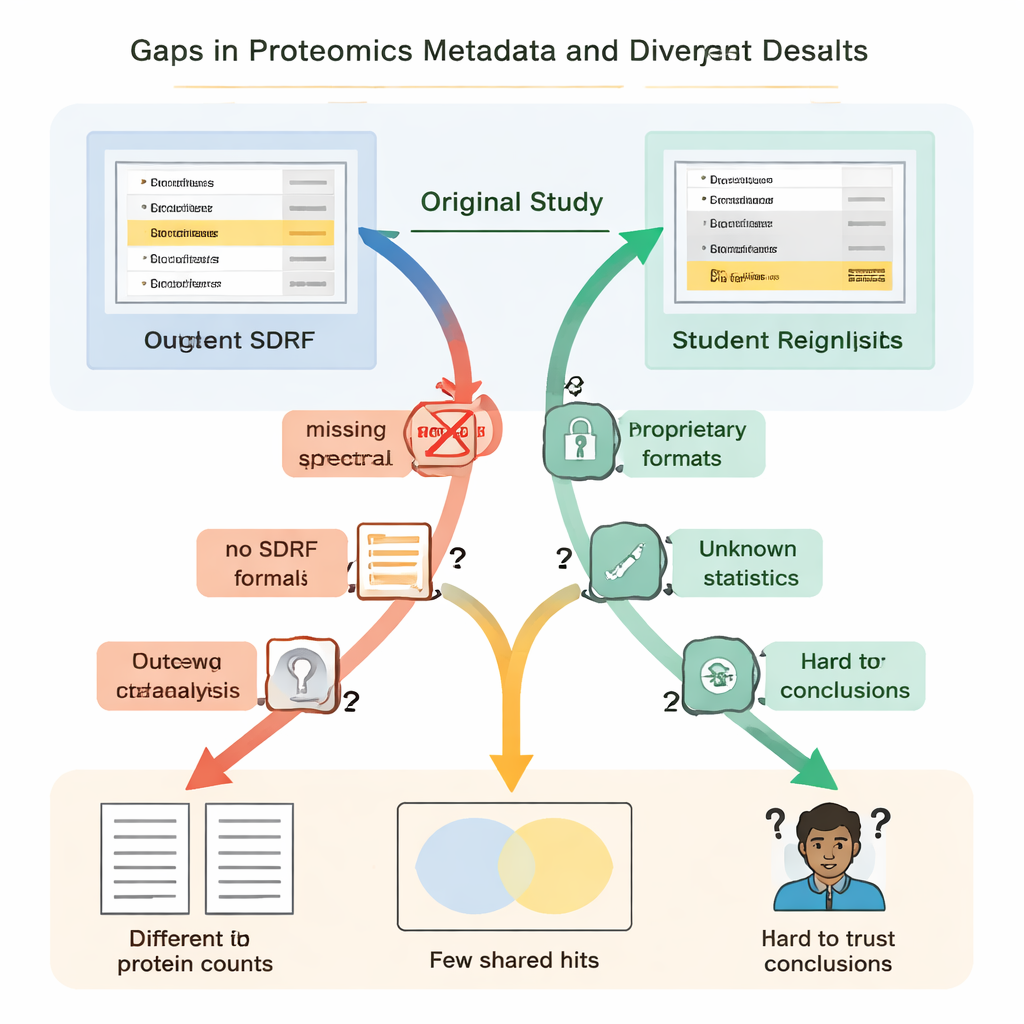
The students quickly discovered that “open” did not always mean “reusable.” In every case, essential instructions were missing or hard to find. Key links between samples and data files were not described in a simple, machine‑readable format, so teams had to guess which raw files matched which biological groups by reading papers and deciphering file names. Details on how false positives were controlled—such as the use of special “decoy” protein sequences—were absent, making it impossible to rigorously judge how trustworthy the reported protein lists really were. In several projects, the main results were locked inside proprietary file formats or depended on commercial software that the students could not access, forcing them to redo large parts of the analysis from scratch.
When small gaps create big differences
These missing pieces were not just nuisances; they led to dramatically different scientific results. In one kidney disease study, the original authors reported just under five thousand proteins, while the student reanalysis—using an open tool and a home‑built spectral library—found over thirteen thousand. A protein highlighted in the original paper as especially important did not appear convincingly in the underlying identification file and was not detected at all in the students’ workflow. In another case, the original study listed 108 proteins as changing between conditions, but the students, working from the same raw data but with incomplete information on how the original statistics were done, could confidently flag only 11. Elsewhere, the lack of biological replicates in the uploaded files meant that proper statistical testing was simply impossible.
What a “reusable” dataset should really contain
From these six case studies, a clear pattern emerged: the main barriers to reproducibility were not the mass‑spectrometry machines themselves but the way results were packaged and shared. The authors argue that every proteomics dataset should come with a minimal re‑analysis package. This includes raw data plus open, community‑standard result formats; a standardized table that links each sample to its experimental conditions; basic quality‑control summaries; any spectral libraries or protein sequence files needed to repeat the search; and complete analysis parameters and code, ideally stored with versioned software containers. Repositories, journals, and reviewers could help by nudging or requiring submitters to provide this bundle up front, so that others do not have to reconstruct the workflow from scattered hints.
Training scientists while fixing the system
The course itself served a double purpose. For students, it offered a hands‑on way to master complex proteomics methods, statistics, and coding, while revealing how fragile published conclusions can be when documentation is incomplete. For the wider community, the students’ struggles provided a stress test of current data‑sharing practices, highlighting exactly where metadata and analysis records fall short. The authors suggest that similar courses could be run elsewhere, turning classrooms into quality‑control engines that continuously push for clearer, more transparent data.
From data tombs to living resources
In plain terms, the article concludes that many protein datasets now sitting in public repositories are at risk of becoming digital graveyards—expensive experiments whose results cannot be reliably checked or extended. Yet the solution is relatively straightforward: treat metadata, open formats, and sharable code as integral parts of the experiment, not afterthoughts. If researchers, reviewers, and repositories collectively insist on a simple, well‑documented package whenever proteomics data are shared, those datasets can remain “alive”: ready to be reanalyzed, combined with new studies, and used to strengthen the evidence behind biomedical discoveries.
Citation: Vadadokhau, U., Soliman, M., Castillon, L. et al. Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement. Sci Data 13, 287 (2026). https://doi.org/10.1038/s41597-026-06614-8
Keywords: proteomics, data reproducibility, open science, mass spectrometry, research data sharing