Clear Sky Science · en
Vitamin B2 metabolism promotes FSP1 stability to prevent ferroptosis
How a Common Vitamin Helps Cells Decide Between Life and Death
Our cells constantly walk a tightrope between survival and self-destruction. One dramatic form of cellular demise, called ferroptosis, has attracted attention because it can selectively kill cancer cells that resist other treatments. This study reveals that an everyday nutrient—vitamin B2, or riboflavin—quietly tips the balance by stabilizing a key protective protein. Understanding this hidden connection between diet, cell metabolism and cancer cell death could help researchers design smarter therapies and refine future nutritional advice.
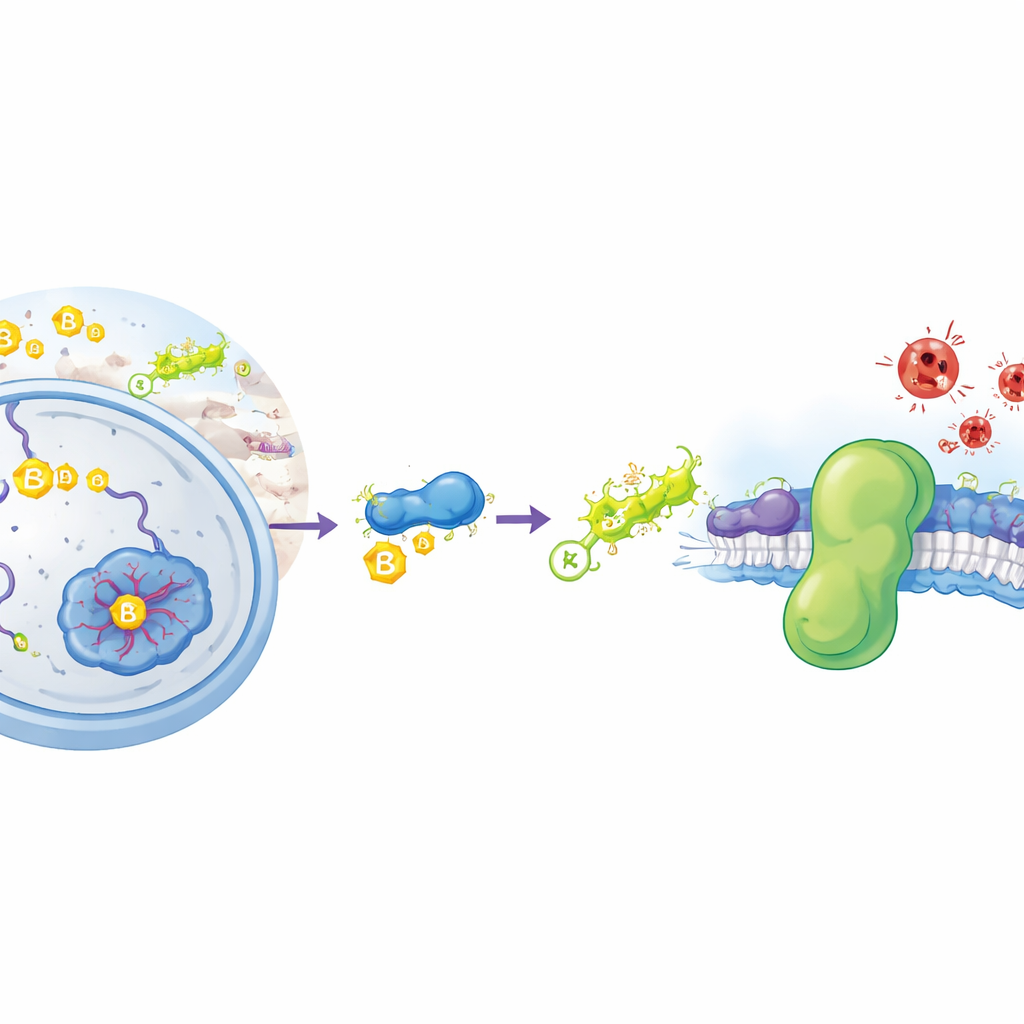
A Special Kind of Rust Inside Cells
Ferroptosis is a type of cell death driven by a chemical process not unlike metal rusting: the uncontrolled oxidation of fats in cell membranes. When these fats break down, membranes lose integrity and cells burst. Our cells normally deploy several defenses to prevent this from happening. One major shield is an enzyme called GPX4, which uses a small molecule, glutathione, to detoxify harmful lipid peroxides. A second, parallel shield is a protein named FSP1, which sits on cellular membranes and uses small fat-like molecules to intercept destructive radicals before they spread. Cancer cells often crank up FSP1 to avoid ferroptosis, making this protein a prime target for new anti-cancer drugs. Yet until now, scientists did not know how cells control how much FSP1 is made and how long it survives.
Building a Cellular “Fuel Gauge” for a Death-Blocker
To uncover the hidden managers of FSP1, the researchers first engineered human bone cancer cells to carry a fluorescent reporter. They tagged the natural FSP1 protein with a green light signal and coupled it to a blue signal that reports how much FSP1 message the cell is making. This clever design let them distinguish changes in gene activity (blue) from changes in protein stability (green). With this dual-color system in place, they used CRISPR–Cas9 to systematically disrupt nearly every gene in the genome and then sorted cells with high or low FSP1 levels. By comparing which guide RNAs were enriched in each group, they mapped hundreds of genes that either boost or diminish FSP1, acting at the level of gene control or protein turnover.
Vitamin B2’s Hidden Job: Making a Stabilizing Handle
Among the most striking hits were two enzymes, riboflavin kinase (RFK) and FAD synthase (FLAD1), which convert vitamin B2 into a cofactor called FAD. FSP1 is a flavoprotein that normally binds FAD tightly to carry out its chemical reactions. When RFK or FLAD1 were removed, or when cells were grown in vitamin-B2-deficient medium, FSP1 protein levels dropped sharply even though its gene activity stayed similar. The team showed that this loss made cells much more vulnerable to ferroptosis when GPX4 was blocked. Importantly, vitamin B2 itself did not act as a classic antioxidant: in a sensitive test tube assay it failed to stop lipid oxidation, unlike vitamin E. Instead, adding FAD (and partly its precursor FMN) to deficient cells restored both FSP1 levels and resistance to ferroptotic death, whereas extra vitamin B2 alone did not help if the processing enzymes were missing.
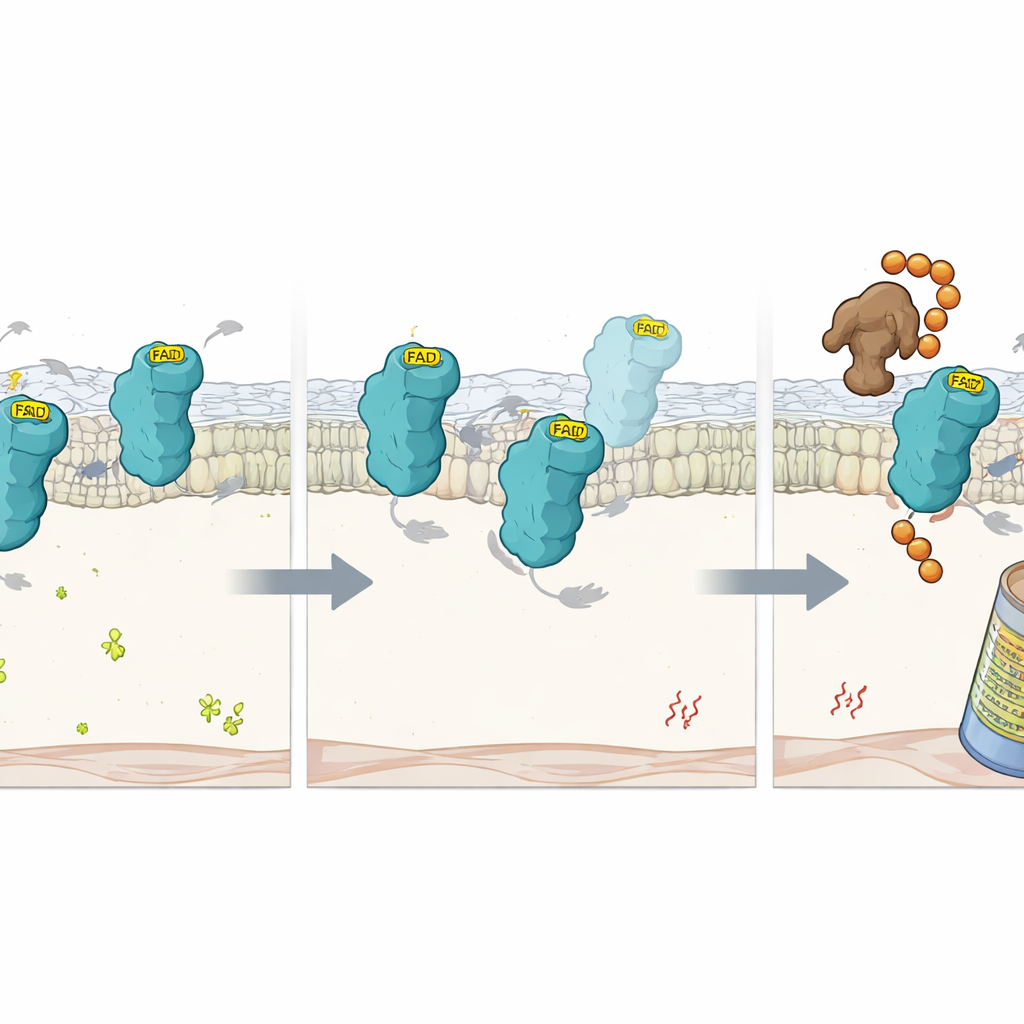
What Happens When the Cofactor Is Missing
To zoom in further, the scientists studied precise mutations in FSP1 that cripple its ability to hold FAD. These mutant proteins still folded into largely normal shapes but lost their FAD and catalytic activity. Inside cells, they decayed far more quickly than normal FSP1, unless the proteasome—the cell’s protein-shredding machinery—was blocked. This suggested that FAD binding itself acts like a stabilizing handle that protects FSP1 from being tagged as defective. Using another focused CRISPR screen under conditions of low FAD, the team identified an E3 ligase called RNF8 as a key factor that recognizes FAD-free FSP1. When RFK was deleted, RNF8 attached chains of ubiquitin tags to the empty protein, targeting it for destruction by the proteasome. Removing RNF8 slowed FSP1 turnover in FAD-poor cells, although it could not restore its lost protective function without the cofactor.
From Molecular Circuitry to Cancer Therapy Ideas
Putting these pieces together, the authors propose a simple but powerful model. Vitamin B2, after being converted into FAD by RFK and FLAD1, binds to FSP1 and is essential both for its biochemical activity and its longevity. When vitamin B2 supply or its processing falters, newly made FSP1 cannot secure FAD, is flagged by RNF8 and is rapidly dismantled by the proteasome, leaving cells more exposed to ferroptotic damage. Cancer data suggest that tumors with higher RFK expression are more resistant to ferroptosis-inducing drugs, underscoring the real-world relevance of this pathway. For non-specialists, the key message is that a familiar vitamin does far more than act as a simple antioxidant: it helps decide whether a potent anti-death protein stands guard or is swept away. By tuning vitamin B2 metabolism or the stability of FSP1, future treatments might better harness ferroptosis to eliminate cancer cells while sparing healthy tissue.
Citation: Deol, K.K., Harris, C.A., Tomlinson, S.J. et al. Vitamin B2 metabolism promotes FSP1 stability to prevent ferroptosis. Nat Struct Mol Biol 33, 525–536 (2026). https://doi.org/10.1038/s41594-026-01759-x
Keywords: ferroptosis, vitamin B2, FSP1, cancer cell death, cell metabolism