Clear Sky Science · en
AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2
Finding Drug Targets in a Sea of Proteins
Modern medicines often work by latching onto tiny nooks and crannies on the surfaces of proteins inside our cells. Yet even with today’s enormous catalogs of protein structures, it remains surprisingly hard to tell in advance where a small molecule – a potential drug – might actually stick. This study introduces AF2BIND, a simple but powerful computational tool that mines the internal workings of AlphaFold2, the landmark protein structure predictor, to spotlight likely drug-binding sites across thousands of human proteins. Its goal is to narrow the search for new medicines and reveal hidden functional hot spots that traditional methods overlook.
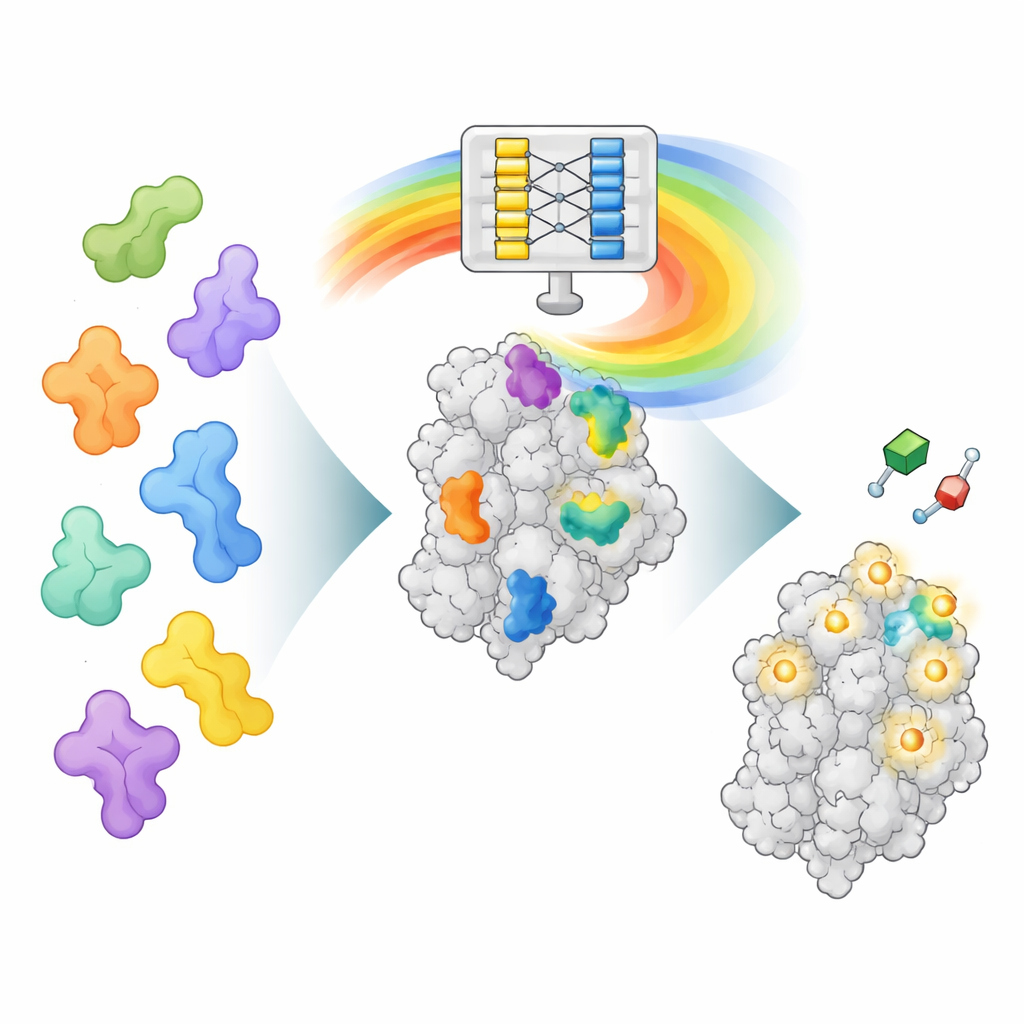
A New Way to Read AlphaFold’s “Mind”
AlphaFold2 was trained to predict how a chain of amino acids folds into a three-dimensional protein, not to find where drugs bind. However, in learning to fold proteins, it also learned rich patterns about how different parts of proteins interact. AF2BIND taps into one of these internal data layers, called the pair representation, which encodes how every pair of amino-acid positions relates in space. The authors feed AlphaFold2 a protein sequence together with its backbone structure and also append 20 extra amino acids, one of each type, as separate “bait” chains. AlphaFold2 then computes how the protein interacts with each bait residue. These interaction patterns become the input to a very straightforward logistic regression model that estimates, for every position in the protein, the probability that it belongs to a small-molecule binding site.
Turning Hidden Signals into Practical Predictions
Training AF2BIND required a carefully curated set of about 1,900 protein–ligand structures where small molecules were bound with high-quality experimental evidence. The researchers went to considerable lengths to avoid “cheating” by similarity: they split their data so that test proteins did not share overall fold, sequence, or even binding-pocket shape with those used for training. On this rigorous benchmark, the AF2 pair representation outperformed several alternative neural-network embeddings, including those based only on sequence or on structure-conditioned sequence design. Using the pair features alone, AF2BIND recovered about two-thirds of known binding residues in the top-ranked predictions and showed strong performance across standard classification metrics, while remaining robust to modest changes in protein shape and side-chain orientations.
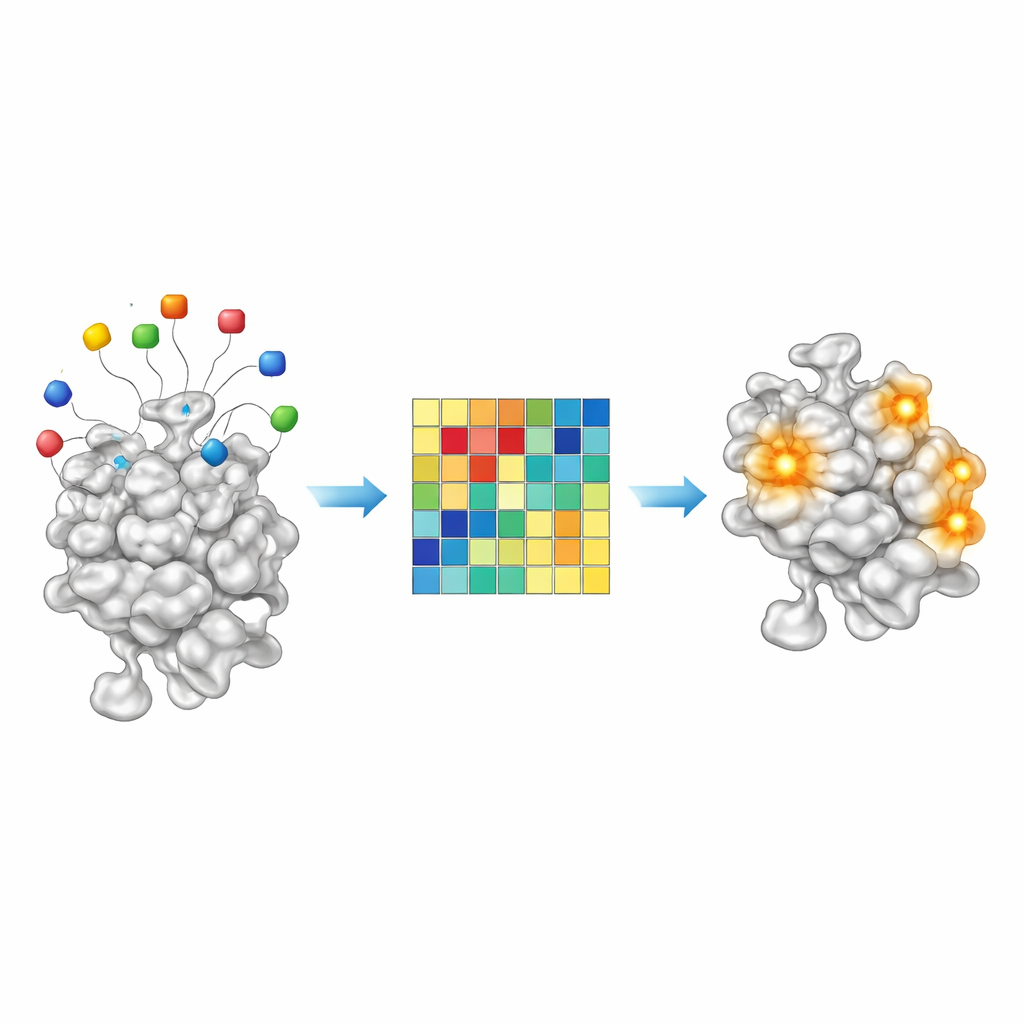
Reading Chemical Clues from Bait Residues
Because AF2BIND is a simple linear model, its decisions are unusually transparent for a modern AI system. Each of the 20 bait amino acids contributes a measurable amount to the final binding score at a given protein position. By examining these contributions across roughly 2,000 protein–ligand complexes, the authors found that certain bait combinations switch on more strongly for oily, carbon-rich ligands, whereas others light up for more polar, water-loving molecules. In other words, the pattern of bait activation acts like a crude chemical fingerprint of what kinds of small molecules a given pocket prefers. This suggests that in the future, AF2BIND-like approaches might not only flag where a drug could bind, but also hint at the sort of chemistry that would fit best.
Scanning the Human Proteome for New Pockets
Armed with their trained model, the team then turned AF2BIND loose on the AlphaFold-predicted structures of the entire human proteome. After trimming away low-confidence regions and splitting very large proteins into manageable structural chunks, they clustered nearby high-scoring residues into candidate binding sites. AF2BIND predicted over 20,000 such sites in more than 13,000 proteins. Strikingly, the majority of these did not overlap with pockets inferred by homology-based methods such as AlphaFill, which copy ligands from related crystal structures, nor with a widely used pocket finder called P2Rank. Many AF2BIND-only sites are shallower or more diffuse than classic buried pockets and often coincide with regions that bind peptides, RNA, DNA, or other proteins—interfaces that may nonetheless be targetable by small molecules.
Implications for Drug Discovery and Disease
To judge how promising these newly suggested sites might be for drug design, the authors used an independent tool that scores “druggability” based on pocket size, enclosure, and chemical environment. On average, AF2BIND’s sites scored above a common threshold for attractive drug targets, including those found in proteins linked to inherited diseases. When cross-referenced with chemoproteomic experiments that label reactive cysteines in cells, AF2BIND and P2Rank together explained nearly half of the observed ligandable regions, each method catching cases the other missed. The work shows that the internal representations learned by structure-prediction networks can be repurposed to map likely drug-binding sites at massive scale, without prior knowledge of any specific ligand. For nonspecialists, the key message is that the same AI breakthroughs that predict protein shapes are beginning to reveal where and how medicines might best grip those shapes, potentially accelerating the search for new treatments and illuminating previously hidden control points in our proteins.
Citation: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Keywords: protein binding sites, drug discovery, AlphaFold2, computational biology, structural bioinformatics