Clear Sky Science · en
DECODE: deep learning-based common deconvolution framework for various omics data
Why this research matters
Modern biomedicine is awash in measurements of our tissues: which genes are active, which proteins are present, and which small molecules fuel our cells. Yet most of these measurements are taken on blended samples, where many cell types are mixed together. The study behind DECODE introduces a powerful artificial‑intelligence framework that can unmix these signals, telling us which cells and cell states are present, even across very different kinds of data. This ability could accelerate research on cancer, immunity and metabolic disease while making better use of existing biobank samples.
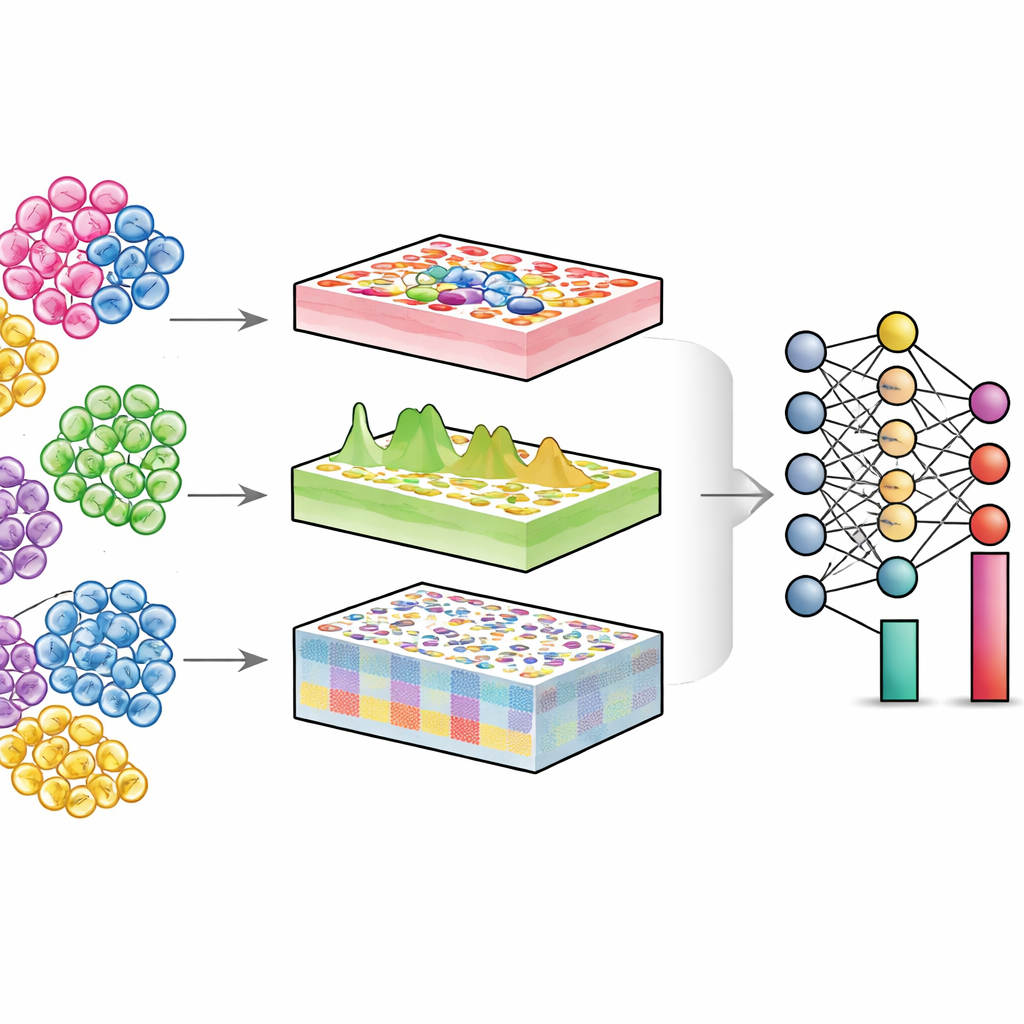
Peeking inside mixed tissues
Every organ is a community of different cell types—immune cells, structural cells, stem cells and more. In health and disease, what often changes is not just what each cell does, but how many of each kind are present and what state they are in. Single‑cell technologies can measure individual cells directly, but they are expensive and technically demanding, especially for large patient cohorts or old stored samples. By contrast, conventional “bulk” experiments mix thousands or millions of cells together and read out an average signal. Deconvolution algorithms attempt to reverse this mixing: given bulk data and a reference map of single cells, they estimate the proportion of each cell type in the tissue.
The limits of one‑trick tools
Existing deconvolution tools are mostly tailored to a single kind of measurement, such as gene activity (transcriptomics) or proteins (proteomics). They often assume specific statistical behaviors that do not hold for other data types, and they struggle when the bulk tissue contains cell types that are missing from the reference data. Strong batch effects—differences in donors, instruments or health states—can further blur the biological signals. Notably, there was no practical method for metabolomics, the study of small molecules that are often closest to clinical symptoms. As a result, scientists analyzing multiomics cohorts had to juggle several specialized tools, each with its own quirks, making it hard to compare results across studies and data types.
A universal unmixing engine
DECODE tackles these challenges by treating deconvolution as a flexible deep‑learning problem that can handle genes, proteins and metabolites in a unified way. First, it synthesizes “pseudotissues” by digitally mixing single‑cell profiles in random proportions, creating a rich training set where the true cell composition is known. An adversarial learning stage then teaches an encoder to map both real tissues and pseudotissues into a shared representation where technical differences are minimized but biologically meaningful patterns are preserved. Next, a special denoising module, guided by contrastive learning, learns to separate true tissue signals from artificial noise. This step makes DECODE robust to missing cell types in the reference data and to measurement errors. Finally, the cleaned features are passed to a deconvolution module that estimates either absolute or relative abundances of cell types and cell states, depending on how complete the reference is.
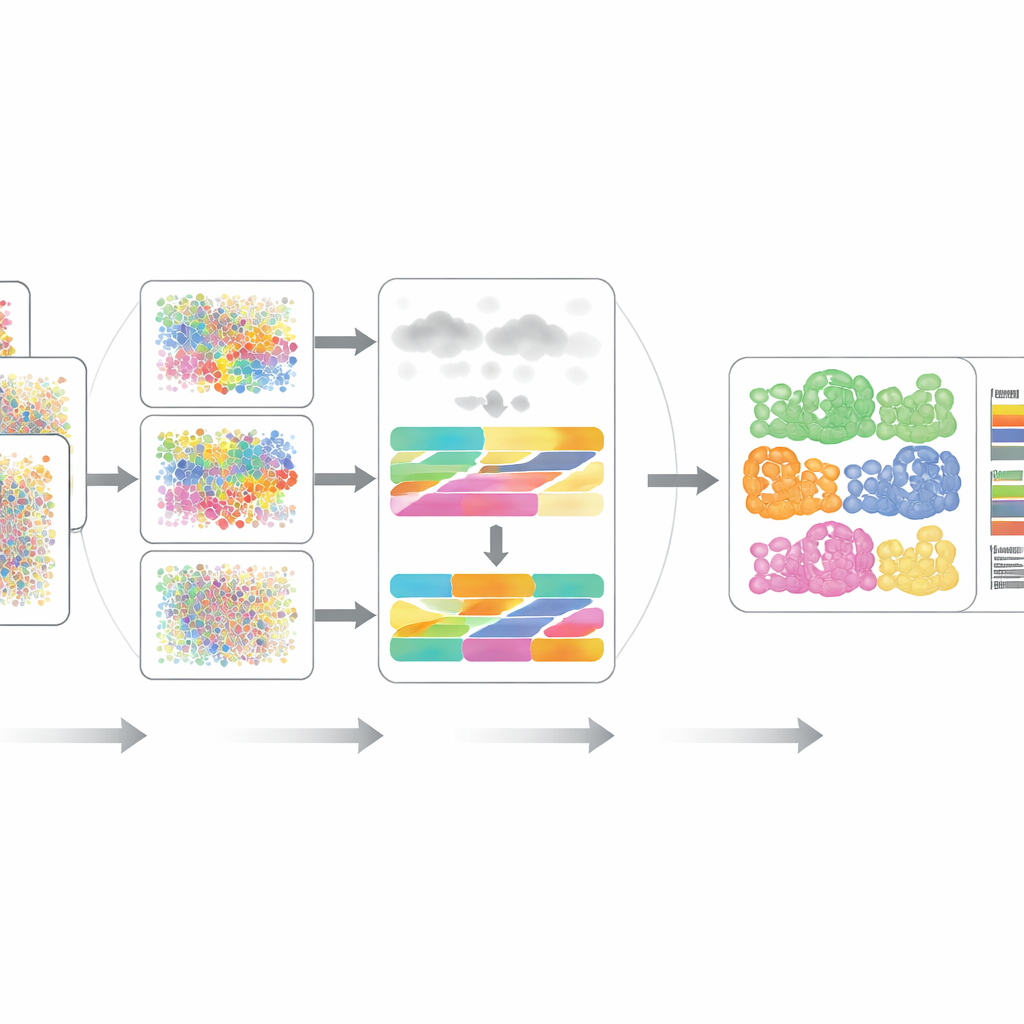
Putting DECODE to the test
The authors rigorously benchmarked DECODE on 15 datasets spanning seven realistic scenarios, including different donors, disease states, health conditions, experimental platforms and even spatially resolved measurements. Across transcriptomics and proteomics, DECODE generally matched or exceeded state‑of‑the‑art tools in accuracy while maintaining reasonable computing time and memory use. Crucially, DECODE was the only method to deliver reliable results on metabolomics data, where there are fewer features and different cell types can look deceptively similar. The framework also proved adept at tracking cell states—such as progression along a developmental trajectory, phases of the cell cycle, or responses to drug treatment—rather than just static cell types.
Robust in noisy and incomplete real‑world data
Real tissues often contain cell types not captured in lab‑based single‑cell references, and experimental noise can distort many features at once. The researchers simulated these problems by adding unknown cell types and introducing several flavors of noise and missing data across transcriptomics, proteomics and metabolomics. In most settings, DECODE remained the most accurate method and, in metabolomics, the only one that did not break down. They also demonstrated that DECODE gives highly consistent answers when applied to matched gene and protein measurements from the same blood‑cell samples, a key requirement for comparing cell‑type changes across omics layers in large cohorts.
New biological insights from multiomics cohorts
Armed with this unified tool, the team revisited complex disease datasets. In breast cancer, they combined transcriptomic and proteomic cohorts to show how immune cells and supportive stromal cells shift between non‑metastatic tumors, metastasizing primary tumors and brain metastases. Patterns such as higher T‑cell and perivascular‑like cell abundance in non‑metastatic lesions, and increased B cells in advanced disease, align with and extend previous biological studies. In mouse liver, DECODE integrated transcriptomic, proteomic and metabolomic cohorts to track how hepatocytes, endothelial cells and resident immune cells change under different diets and liver‑disease models, recapitulating known trends such as rising Kupffer cell fractions in inflammatory conditions.
What this means going forward
For a lay reader, the main message is that DECODE acts like a smart prism for biomedical data: given blended measurements from tissues, it can separate out the contributions of many different cell types and states, and it does so reliably across several kinds of molecular readout. This enables scientists to squeeze much more information out of existing multiomics cohorts and biobanks without collecting new single‑cell data for every project. While the method still depends on the quality and breadth of available single‑cell references, and metabolomics resources remain limited, DECODE marks a significant step toward routine, cell‑level interpretation of large‑scale human studies, with potential benefits for understanding disease mechanisms and guiding precision medicine.
Citation: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Keywords: multiomics deconvolution, single-cell reference, deep learning in biology, metabolomics analysis, cell-type composition