Clear Sky Science · en
Rate variation and recurrent sequence errors in pandemic-scale phylogenetics
Why this matters for future outbreaks
When a new virus spreads around the globe, scientists race to read its genetic code and reconstruct its family tree. Those trees help track how variants arise, how fast they spread and whether control measures work. But during COVID-19, labs sequenced millions of SARS‑CoV‑2 genomes so quickly that hidden errors and quirks in the data began to distort the picture. This paper introduces new methods to clean and interpret such vast genetic datasets, offering clearer views of how a pandemic virus really evolves and moves through populations.
The challenge of making sense of millions of genomes
Genomic epidemiology turns virus genomes into practical information for public health decisions. For SARS‑CoV‑2, more than 20 million genomes have been shared worldwide. Traditional evolutionary tools were built for more modest problems, such as comparing genes between species, not handling millions of near‑identical viral sequences arriving in real time. On this scale, two issues become especially troublesome. First, some sites in the viral genome mutate far more often than others, which can make unrelated viruses look oddly similar. Second, recurring technical errors in sequencing and data processing can mimic real mutations. Both effects generate “false echoes” in the evolutionary tree, creating uncertainty about which branches and groupings to trust.
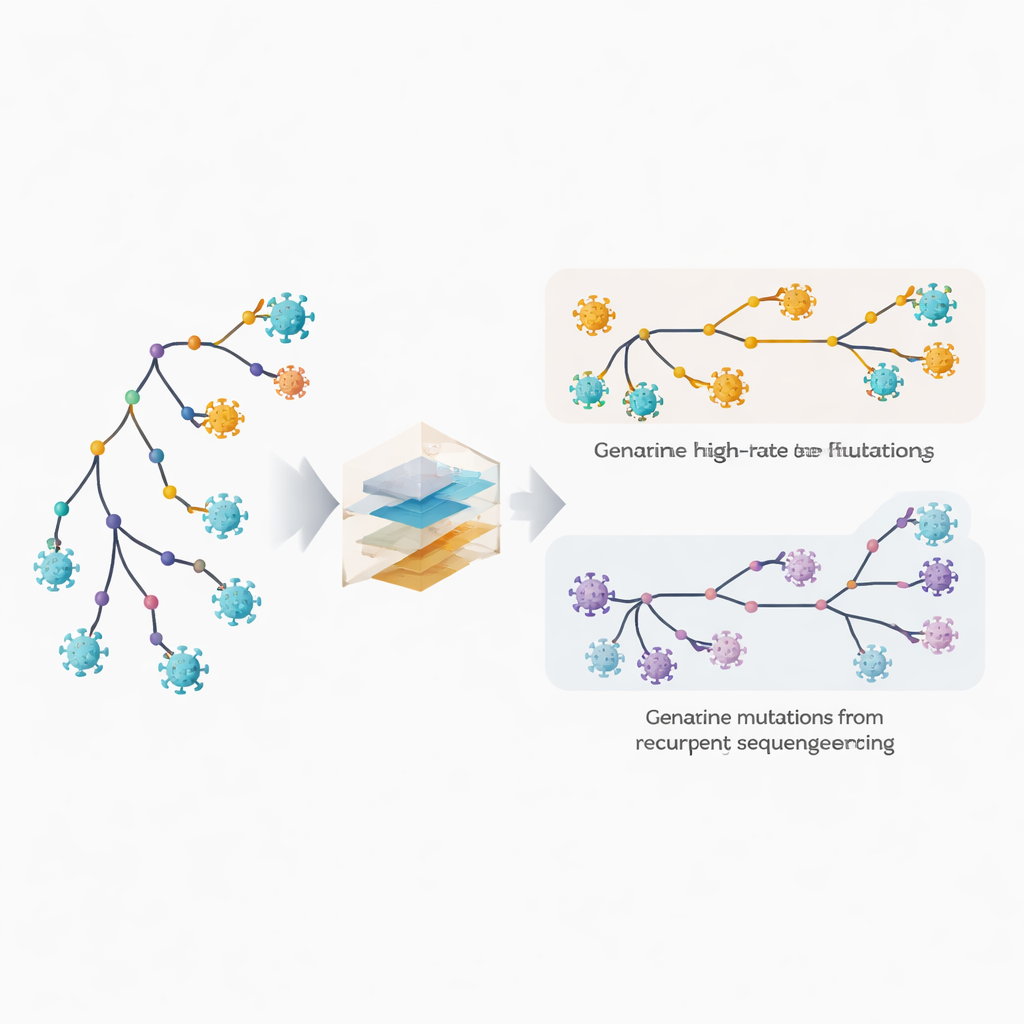
Spotting fast‑changing sites and hidden mistakes
The authors extend their phylogenetic software, MAPLE, with models that treat each position in the viral genome as having its own behavior. Instead of assuming a handful of average mutation rates, the method estimates a separate rate for every site, taking advantage of the huge number of available genomes. At the same time, it allows each site to have its own probability of carrying a recurring sequencing or consensus‑calling error. The key trick is to compare how often a change appears on deep internal branches of the tree, which reflect older, shared events, versus on the outermost tips, which correspond to individual genomes. True biological mutations tend to be spread between internal and terminal branches, whereas technical errors show up mostly at the tips. By exploiting this pattern, the method can disentangle genuine rapid evolution from repeated mistakes.
Faster algorithms for a crowded tree of life
Handling millions of genomes would normally require enormous computing power. To keep the analysis practical, the team redesigned how MAPLE stores and updates sequence information on the tree. Instead of comparing every genome to a single fixed reference, the software selects “local reference” points inside the tree and records nearby genomes as differences relative to these anchors. This compact representation speeds up comparisons between distant parts of the tree. Additional improvements refine how new samples are added to an existing tree, how branch lengths are tuned, and how likely alternative tree shapes are explored, with options to run the most demanding steps in parallel across multiple processor cores.
Testing the method and cleaning real‑world data
To check that their models work, the authors first created realistic simulated SARS‑CoV‑2 datasets with known mutation patterns and embedded sequence errors. On these tests, the new approach recovered truer evolutionary trees and located individual errors with high precision, especially when tens of thousands of genomes or more were included. They then turned to real data, analyzing millions of SARS‑CoV‑2 sequences for which raw reads were available. By comparing two different consensus‑building pipelines, they pinpointed specific genome positions repeatedly affected by artifacts, such as primer binding problems or reference‑biased calling. These suspect sites were masked from further analysis, and genomes showing signs of contamination or mixed infection were filtered out, yielding a curated alignment of over two million high‑quality sequences.
A clearer global picture of the virus family tree
Using the cleaned dataset, the authors reconstructed a global SARS‑CoV‑2 phylogenetic tree and mapped how major variants relate to each other. Their tree sometimes proposes subtly different relationships than previous public trees, often in ways that require fewer mutation events and better match the statistical model. The framework also highlights where lineage labels may be inconsistent with the underlying genetic history, flagging possible recombinants or problematic genomes for closer inspection. Although some challenges remain—such as over‑fitting when data are scarce, or the influence of heavily contaminated samples—the work shows that it is now feasible to build more reliable, pandemic‑scale evolutionary trees. For a lay reader, the bottom line is that better handling of errors and mutation hot spots leads to sharper insight into how pathogens spread and change, helping scientists and health agencies respond more quickly and confidently in future outbreaks.
Citation: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Keywords: SARS-CoV-2 genomics, phylogenetic methods, sequencing errors, mutation rate variation, genomic epidemiology